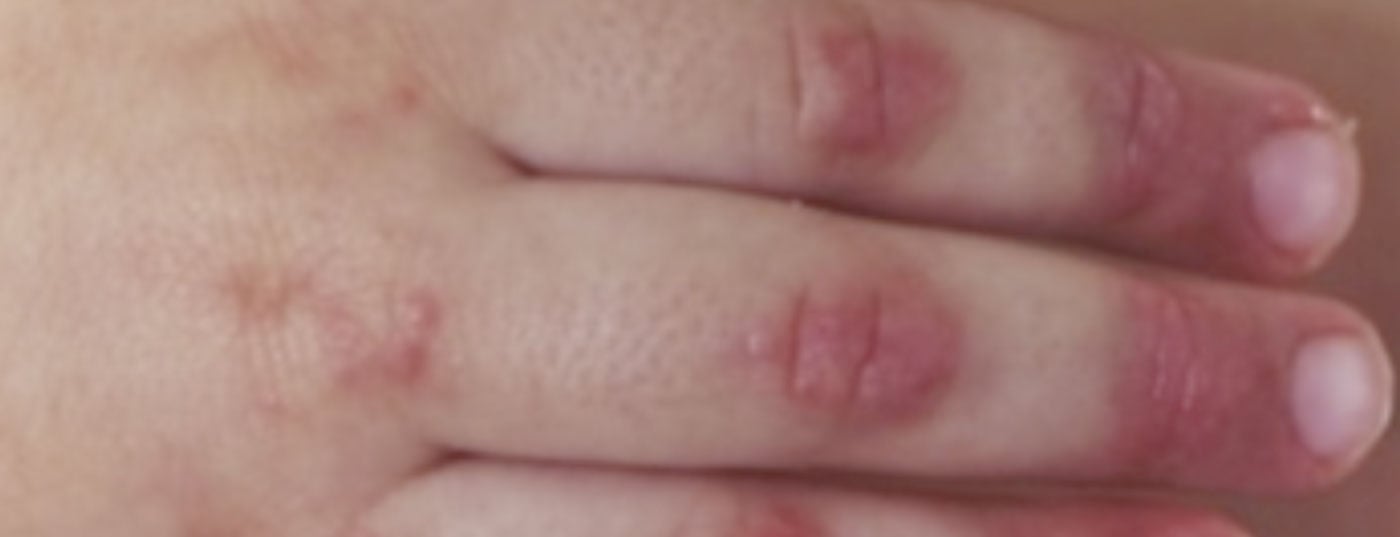
Um tratamento precoce e adequado pode ter um efeito positivo no curso futuro desta doença multissistémica rara. Este artigo dá uma breve visão geral do que é conhecido hoje. Entre outras coisas, a identificação de novos biomarcadores poderia servir de base para uma terapia personalizada no futuro.
A dermatomiosite juvenil (JDM) é uma doença sistémica, imuno-mediada da infância, caracterizada por fraqueza muscular e erupções cutâneas típicas [9]. Outros sistemas orgânicos, tais como o coração e os pulmões, também podem ser afectados e continuam a ser a causa de morte mais comum nestes pacientes. É a miopatia inflamatória idiopática mais comum na infância, com uma incidência de 2-4/milhões/ano.
O processo inflamatório é caracterizado pela infiltração do tecido afectado por células do sistema imunitário, tais como células T e células plasmocitóides dendríticas, e uma assinatura de interferão [1]. A maioria dos sintomas deve-se à vasculopatia, que resulta na falta de funcionamento e perda de células endoteliais. A imunossupressão eficaz, geralmente uma terapia combinada de metotrexato subcutâneo e corticosteróides orais, reduziu consideravelmente a mortalidade (actualmente em cerca de 1%) e cerca de 50% dos doentes apresentam apenas uma erupção da doença antes que a remissão possa ser alcançada [2]. Os outros 50% mostram um curso crónico, com recaídas frequentes, aumento da morbilidade e redução da qualidade de vida [2].

Subfenotipagem através de biomarcadores
O JDM é uma doença muito heterogénea com fenótipos diferentes, em que diversos sistemas de órgãos podem ser afectados em diferentes graus. Os auto-anticorpos têm sido alvo de investigação intensiva nos últimos anos e têm sido capazes de caracterizar melhor os diversos fenótipos; por conseguinte, os auto-anticorpos têm valor prognóstico [3]. A monitorização da actividade da doença e dos danos de órgãos causados pela inflamação é uma parte importante da gestão destes pacientes jovens, uma vez que determina o resultado a longo prazo desta doença [4]. Vários instrumentos estão à disposição do clínico para o fazer, tais como a histopatologia do músculo e tecido cutâneo, microscopia capilar (Fig. 1), escores clínicos para avaliar o envolvimento do músculo e da pele [5], e agora também vários biomarcadores no sangue [6,7]). Os resultados clínicos utilizados para avaliar a actividade da doença requerem a cooperação do doente, o que por vezes pode ser um desafio para as crianças mais novas. Estes escores clínicos são também de utilidade limitada na detecção de inflamação subjacente ou na diferenciação de outras causas de fraqueza muscular, tais como a toxicidade dos esteróides.

Directrizes para a optimização da gestão de doenças
Em 2012, a iniciativa europeia SHARE (Single Hub and Access point for pediatric Rheumatology in Europe) foi lançada com o objectivo de padronizar o diagnóstico e a gestão terapêutica para crianças e adolescentes com doenças reumáticas em toda a Europa. Para o JDM, estas recomendações foram publicadas em 2017 [6].
Diagnóstico: O diagnóstico de dermatomiosite juvenil continua a ser principalmente clínico, com resultados cutâneos típicos (Fig. 2) e com uma debilidade muscular acentuada a curto prazo. Por conseguinte, se houver suspeita de JDM, deve ser feito um encaminhamento rápido para um centro de excelência próximo. Uma terapia precoce e adequada pode prevenir danos a longo prazo e aumentar a possibilidade de remissão precoce. Os critérios clássicos de diagnóstico de Bohan e Peter de 1975 contêm cinco elementos:
- sinais cutâneos característicos
- Fraqueza muscular proximal
- biópsia muscular patológica típica
- enzimas musculares elevadas
- alterações miopáticas no electromiograma (EMG)
→ Na prática reumatológica pediátrica, o EMG foi substituído por RM não dolorosa (Fig. 3) [8].
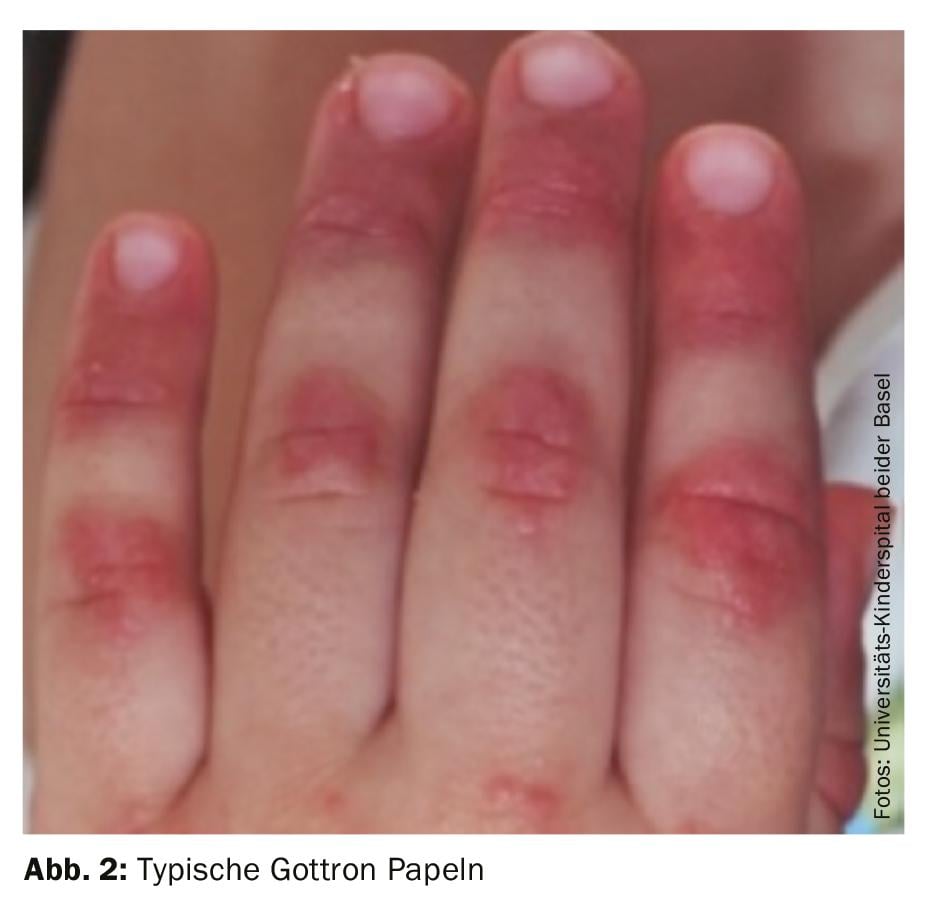

Foram publicados novos critérios de classificação em 2017; por exemplo, omitem biopsia muscular para sintomas patogénicos da pele e também incluem auto-anticorpos (anti-Jo-1) nos critérios [9]. O ultra-som é também cada vez mais utilizado, uma vez que não requer sedação nas crianças mais novas.
A fraqueza muscular deve ser avaliada na criança com diferentes instrumentos. Estão disponíveis testes musculares funcionais, o chamado “Children’s Myositis Activity Score” (CMAS) [10], bem como testes musculares que medem apenas a força, o chamado Teste Muscular Manual (MMT) [11]. Ambos os testes são úteis na avaliação da fraqueza muscular no diagnóstico; mas também servem como importantes parâmetros de acompanhamento que podem dar uma indicação significativa da actividade da doença. Ambos os testes fazem parte dos critérios de remissão de 2013 [12].
Wienke et al. validaram recentemente vários biomarcadores (galectina-9 e CXCL10) que desempenham um papel não só no diagnóstico mas também no acompanhamento e podem também facilitar as decisões de tratamento no início da doença: Neste estudo prospectivo, os pacientes com níveis elevados destes biomarcadores no momento do diagnóstico mostraram um curso mais severo no seguimento e poderiam, portanto, beneficiar potencialmente de uma terapia mais intensiva. Em contraste, em pacientes com apenas uma recidiva, os valores já tinham quase normalizado alguns meses após o diagnóstico sob terapia. Além disso, foi possível demonstrar que os marcadores já mostravam um aumento renovado do sangue antes das descobertas clínicas de uma crise de doença [7].

Terapia 2: A terapia de primeira linha continua a ser uma combinação de esteróides e eu-trexate, possivelmente ciclosporina [6]. O metotrexato deve ser mantido durante pelo menos 2 anos; os esteróides são descontinuados no 1º ano de terapia no caso de um curso monofásico [6]. Se certos sintomas como fraqueza muscular grave (a criança já não consegue sair da cama, disfagia grave), deficiência da função miocárdica ou pulmonar ou úlceras cutâneas indicarem um curso grave, a ciclofosfamida deve ser adicionada. Se não houver melhoria, devem ser adicionadas imunoglobulinas intravenosas ou ciclosporina A, ou mudar para biólogos (rituximab, infliximab, adalimumab). De um ponto de vista pediátrico, o aumento da calcinose do tecido subcutâneo ou muscular (Figs. 3 e 4) é um sinal de inflamação sistémica persistente, o que justifica a intensificação da terapia. Os transplantes de células estaminais também têm sido realizados com sucesso em pacientes JDM com um curso muito severo; alguns pacientes conseguiram uma remissão completa sem terapia. Quando a remissão clínica é alcançada, a calcinose pode ser completamente resolvida.
Com bom sucesso, esta terapia padrão permanece associada ao risco de efeitos secundários indesejáveis, tais como infecções, fraqueza muscular induzida por esteróides ou náusea semanal sob metotrexato. No futuro, os novos biomarcadores acima mencionados poderão permitir a identificação precoce de uma nova crise de doença ou uma redução mais rápida da terapia. Os biomarcadores poderiam assim apoiar o caminho para uma terapia personalizada para esta doença rara.
| Ferramenta de diagnóstico baseada na Web Entretanto, está disponível uma ferramenta web que, para além do diagnóstico, dá também uma probabilidade do diagnóstico com a classificação do subgrupo. Estes critérios foram validados tanto para crianças como para adultos com suspeita de miopatia inflamatória. http://www.imm.ki.se/biostatistics/calculators/iim/ |
Literatura:
- Piper CJM, et al: CD19(+)CD24(hi)CD38(hi) B Cells Are Expanded in Juvenile Dermatomyositis and Exhibit a Pro-Inflammatory Phenotype After Activation Through Toll-Like Receptor 7 and Interferon-alpha. Immunol frontal 2018; 9: 1372.
- Gowdie PJ, Allen RC, Kornberg AJ, Akikusa JD: Características clínicas e curso da doença de pacientes com dermatomiosite juvenil. Int J Rheum Dis 2013; 16(5): 561-567.
- Rider LG, Nistala K: As miopatias inflamatórias idiopáticas juvenis: patogénese, fenótipos clínicos e de autoanticorpos, e resultados. J Intern Med 2016; 280(1): 24-38.
- Huber A, Feldman BM: Resultados a longo prazo na dermatomiosite juvenil: como chegámos aqui e para onde vamos? Curr Rheumatol Rep 2005; 7(6): 441-446.
- Huber AM, et al: Validação preliminar e significado clínico da Ferramenta de Avaliação Cutânea na dermatomiosite juvenil. Arthritis Rheum 2008; 59(2): 214-221.
- Enders FB, et al: Recomendações baseadas no consenso para a gestão da dermatomiosite juvenil. Ann Rheum Dis 2017; 76(2): 329-340.
- Wienke J, et al: Galectin-9 e CXCL10 como Biomarcadores da Actividade da Dermatomiosite Juvenil: Um Estudo de Coorte Longitudinal e Validação Multicohort. Arthritis Rheumatol 2019. doi.org/10.1002/art.40881
- Brown VE, et al: Um estudo de consenso internacional sobre os critérios de diagnóstico da dermatomiosite juvenil (JDM). Rheumatology (Oxford) 2006; 45(8): 990-993.
- Lundberg IE, et al.: 2017 European League Against Rheumatism/American College of Rheumatology classification criteria for adult and juvenile idiopathic inflammatory myopathies and their major subgroups. Ann Rheum Dis 2017; 76(12): 1955-1964.
- Huber AM, et al: Validação e significado clínico da Escala de Avaliação da Myosite Infantil para avaliação da função muscular nas miopatias inflamatórias idiopáticas juvenis. Arthritis Rheum 2004; 50(5): 1595-1603.
- Jain M, et al: Fiabilidade intra e inter-rater do Teste Muscular Manual de 10 pontos (MMT) de força em crianças com miopatias inflamatórias idiopáticas juvenis (JIIM). Phys Occup Ther Pediatr 2006; 26(3): 5-17.
- Lazarevic D, et al: Os critérios PRINTO para doença clinicamente inactiva na dermatomiosite juvenil. Ann Rheum Dis 2013; 72(5): 686-693.
- Mathiesen PR, et al: Aptidão aeróbica após o JDM – um estudo de acompanhamento a longo prazo. Reumatologia (Oxford) 2013; 52(2): 287-295.
PRÁTICA DA DERMATOLOGIA 2019; 29(4): 26-29