As terapias inovadoras têm melhorado o prognóstico de muitas doenças oncológicas. Uma importante limitação para o prognóstico individual são as complicações cardiovasculares associadas à doença e à terapia. Mesmo acontecimentos raros podem ter consequências graves.
As terapias inovadoras têm melhorado significativamente o prognóstico de muitas doenças oncológicas nos últimos anos. Uma importante limitação para o prognóstico individual após uma terapia oncológica bem sucedida são as complicações cardiovasculares associadas à doença e à terapia. Apesar dos efeitos cardiotóxicos das terapias oncológicas, algumas das quais conhecidas há muito tempo, existem apenas poucos dados sobre estratificação de risco e tratamentos específicos. Isto aplica-se às terapias sistémicas clássicas (por exemplo, antraciclinas, taxanos ou imunomoduladores), mas especialmente às terapias mais recentes (por exemplo, inibidores de pontos de controlo ou inibidores de proteasomas). Utilizando os inibidores BRAF/MEK como exemplo, os desafios terapêuticos da perspectiva da cardio-oncologia serão discutidos no seguinte.
Exemplo de melanoma maligno
O melanoma maligno é frequentemente diagnosticado em homens e mulheres e é, por exemplo, a quarta manifestação mais comum de um novo cancro na Suíça, com base no relatório de 2017 da Liga do Cancro. As mutações em oncogenes estão frequentemente envolvidas causalmente no desenvolvimento de melanomas malignos. As mutações BRAF são encontradas em cerca de metade de todos os melanomas não renováveis ou metastáticos. Até recentemente, os doentes afectados tinham poucas opções de tratamento com taxas de resposta inferiores a 20%. De acordo com a directriz de 2016, recomenda-se portanto a terapia com um inibidor de BRAF/MEK [1].
BRAF é uma serina citoplasmática/trêsonina cinase que desempenha um papel regulador na regulação do crescimento celular dentro da via Ras-Raf-MEK1-ERK1/2. A mutação mais frequente afecta, de longe, o códão 600. 90% das mutações mudam de valina para ácido glutâmico (V600E) [2], resultando numa activação descontrolada. Isto levou ao desenvolvimento de inibidores específicos: vemurafenibe e dabrafenibe, bem como os inibidores da kinase MEK a jusante: trametinibe e cobimetinibe. Em ensaios clínicos, a combinação de preparações de ambos os grupos de substâncias foi capaz de mostrar um benefício adicional significativo no que diz respeito à sobrevivência sem progressão e à sobrevivência global [3]. Por exemplo, em doentes com melanoma metastático não tratado e mutação BRAF V600E ou V600K, foi encontrada uma sobrevida média de 7,3 meses sem progressão com monoterapia de virurafenibe em comparação com 11,4 meses no grupo de terapia combinada com dabrafenibe e trametinib [4]. Só os inibidores MEK, como mostrado para o trametinibe, mostraram anteriormente melhorar a sobrevivência sem progressão mediana para 4,8 meses em comparação com 1,5 meses como o ponto final primário em comparação com a quimioterapia convencional. A Figura 1 apresenta uma visão simplificada dos inibidores.

Tornou-se evidente uma série de efeitos adversos dos inibidores do BRAF e do MEK. O Vemurafenibe mostrou principalmente artralgia, erupção cutânea, fadiga, alopecia, fotossensibilidade, náusea, queratoacantoma ou carcinoma de células escamosas no ensaio de fase III BRIM-3 [5]. O dabrafenibe mostrou manifestações cutâneas em particular, comparáveis aos efeitos adversos relatados do vemurafenibe, sendo a febre vista como uma complicação mais frequente nos doentes tratados com dabrafenibe [6].
Dados clínicos sobre os efeitos secundários cardíacos
Do ponto de vista clínico, por um lado, a função cardíaca, representada pela fracção de ejecção do ventrículo esquerdo (FEVE), e por outro lado, as arritmias potencialmente malignas (taquicardia ventricular, fibrilação ventricular) recém ocorridas, são relevantes para o prognóstico individual. Em estudos clínicos, um prolongamento do tempo de QT é considerado um indicador da potencial ocorrência de arritmias malignas.
Foi observada uma redução na LVEF em terapia combinada (dabrafenib e trametinib) em 8% dos pacientes, em 3% dos pacientes isto levou à interrupção do estudo. Metade dos doentes afectados (cerca de 4%) sofreram um grau mais elevado de comprometimento das FEVE. Não houve diminuição de LVEF no grupo de monoterapia de virurafenibe. No entanto, os pacientes com uma insuficiência cardíaca pré-existente correspondente à NYHA II ou superior foram excluídos do estudo, de modo que os dados não são geralmente transferíveis para outros colectivos [4]. As taxas de interrupção do estudo foram comparáveis em ambos os grupos entre 12% e 13%, tal como a toxicidade global.
Num estudo comparável, 2% dos pacientes do grupo de monoterapia dabrafenibe mostraram uma diminuição da FEVE, com 4% dos pacientes do grupo de terapia combinada (dabrafenibe e trametinibe) a mostrar uma redução da FEVE [7]. A combinação de vemurafenibe e do inibidor MEK cobimetinibe também resultou num aumento das taxas de comprometimento do LVEF (8% vs. 3% no respectivo grupo) em comparação com a monoterapia [8]. Tal como no caso das terapias combinadas descritas com inibidores de BRAF, o agravamento da FEVE ocorreu numa proporção comparável de 7% com a monoterapia de trametinibe, tendo 1% dos doentes tido de interromper o estudo devido ao desenvolvimento de FEVE altamente prejudicada [9].
Outros efeitos secundários cardíacos foram observados no prolongamento do tempo de QT (por exemplo, no estudo fase II BRIM-2). No entanto, não foi encontrada indução de um episódio de ritmo ameaçador de vida [10]. Em coortes maiores, foram encontrados prolongamentos relevantes de QT acima dos 480 ms em 7% dos pacientes, sem evidência de arritmias malignas. Em 0,5% dos casos, o tratamento com vemurafenibe foi descontinuado devido ao prolongamento do QT [11]. Larkin et al. também relatam num grande colectivo (3222 pacientes) tempos de QT prolongados em 10% dos casos, mais de 500 ms em 2% e dois pacientes com arritmias relevantes [12].
Em resumo, os efeitos secundários cardíacos com limitações no LVEF são em grande parte devidos à adição de inibidores de MEK. A extensão dos efeitos adversos cardíacos é comparável apenas na terapia com inibidores MEK e em combinação com a terapia com inibidores BRAF. Por outro lado, a terapia combinada, em particular, é superior em termos de resultado oncológico, especialmente no que diz respeito à sobrevivência sem progressão e ocorrência de segundas neoplasias malignas da pele.
Para além da redução do prolongamento do tempo LVEF e QT, foram descritos aumentos da pressão arterial com uma incidência de 15-25% para as substâncias. Em contraste com a restrição da LVEF ou a ocorrência de um tempo QT prolongado, as medidas terapêuticas com medicamentos anti-hipertensivos comuns, bem como os controlos clínicos através da medição da pressão arterial, são fáceis de derivar em caso de hipertensão.
O que podemos aprender com os modelos pré-clínicos ?
As vias moleculares de sinalização das kinases proteicas mitogénicas (MAP kinases) no cardiomiócito têm sido amplamente estudadas a nível celular e em modelos animais. Com base nisto, a activação da via Ras-Raf-MEK1-ERK1/2 nos cardiomiócitos está associada à hipertrofia celular mediadora e à apoptose. Os membros da cascata de sinalização interagem com uma variedade de outras kinases MAP, tais como C-Jun N-terminal kinase (JNK), p38 e kinase 5 extra-celular regulada pelo sinal (ERK5) [13]. Classicamente, a activação de ERK1/2 está associada a processos de crescimento, enquanto que as kinases MAP JNK e p38 são mais susceptíveis de serem activadas no contexto de estímulos patológicos. A activação artificial da via MEK1-ERK1/2 é, portanto, mais susceptível de ser cardioprotectora, enquanto a activação de outras kinases MAP como a p38 ou JNK é mais susceptível de conduzir a efeitos cardíacos adversos [14].
As mutações na via ERK1/2, nomeadamente a ERK1/2 fosfatase parental PTPN11 e a proteína G K-RAS, estão causalmente associadas a defeitos cardíacos congénitos tais como as síndromes Noonan e LEOPARD. As perturbações cardíacas e defeitos de septo, mas sobretudo as cardiomiopatias hipertróficas, têm sido descritas nestas síndromes.
Inactivação da PTNP11 (mutação Y279C) em fenocópias de ratos a síndrome cárdio-fácio-cutânea, incluindo o desenvolvimento de cardiomiopatia hipertrófica primária, que progride para cardiomiopatia dilatada com o avanço da idade dos animais [15]. No modelo do rato, a cardiomiopatia hipertrófica também poderia ser desencadeada por Raf1-L613V-knock-in, uma mutação da síndrome Noonan humana [16]. Em resposta ao aumento da carga de volume, os ratos mostraram um aumento da formação de fibrose cardíaca intersticial e perivascular e reduziram significativamente a sobrevivência global. A actividade MEK e ERK são mais induzíveis no “knock-in” com ligação ligand, pelo que o fenótipo cardíaco descrito poderia ser evitado através de terapia com um inibidor MEK.
A sobreactivação artificial com sobreexpressão cardíaca de MEK1, por outro lado, leva a uma hipertrofia cardíaca compensada sem, pelo menos nos modelos animais, aumento da letalidade. A LVEF foi mesmo melhorada ecocardiograficamente com função diastólica prejudicada. ERK1/2 foram upregulados no modelo transgénico MEK1 [17]. A activação da via Raf-1/MEK/ERK é também necessária para a hipertrofia cardíaca durante a carga de pressão aumentada [18]. Em processos de remodelação patológicos no contexto de enfarte do miocárdio, a activação da via de sinalização ERK também levou à cardioprotecção.
Em geral, emerge um quadro complexo dos benefícios e efeitos adversos das kinases MAP individuais, com um efeito pronunciado sobre a hipertrofia cardíaca na maioria dos artigos. Alterações consecutivas (apoptose, fibrose, disfunção cardíaca) resultam de indução de stress adicional ou sobreativação de vias de sinalização individuais (tab. 1).
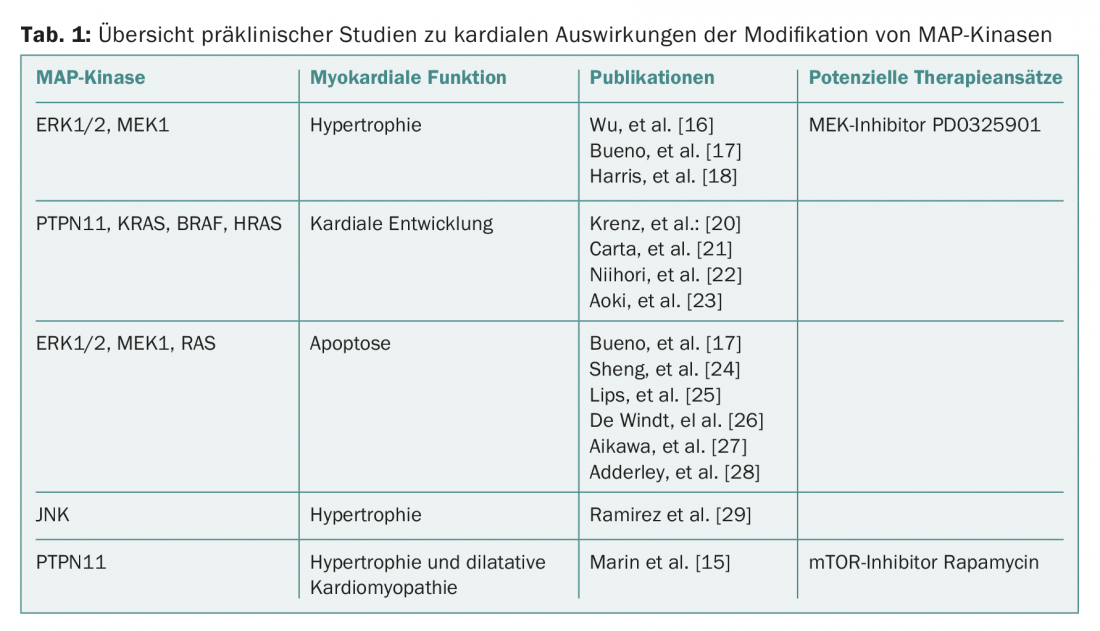
Em parte, as observações clínicas de cardiotoxicidade da inibição de MEK podem ser replicadas. A intervenção selectiva na via de sinalização Ras-Raf-MEK-ERK1/2 ou vias de sinalização associadas – como demonstrado para a inibição de mTOR com rapamicina – poderia ser uma abordagem futura para reduzir os efeitos secundários cardíacos no contexto de terapias oncológicas com inibição de BRAF/MEK. Também se pode assumir que as imagens de última geração (por exemplo, ressonância magnética cardíaca ou ecocardiografia 3D) podem identificar mais cedo os pacientes com activação de vias pró-hipertróficas.
Conclusão para a prática
Os efeitos secundários cardíacos dos inibidores de BRAF/MEK têm sido documentados até agora num número relativamente pequeno de pacientes na faixa percentual de um dígito. No entanto, em caso de ocorrência, estas podem levar a consequências graves e ao término da terapia. A biologia molecular revelou uma imagem diferente das kinases MAP individuais, particularmente no que diz respeito à hipertrofia cardíaca e apoptose. O que não é claro são as condições exactas de stress cardíaco e factores predisponentes de um paciente que levam a um agravamento clinicamente significativo da FEVE durante a terapia. Por esta razão, é necessária uma monitorização cardíaca estreita e eficiente dos pacientes antes e durante a terapia de inibição de BRAF/MEK, para que os pacientes afectados (deterioração da LVEF, prolongamento do tempo QT, hipertensão arterial) possam ser tratados imediatamente pelos cardiologistas. Se necessário, a terapia oncológica pode então ser continuada sob estreita vigilância.
De acordo com as orientações do CES, o tratamento dos efeitos cardiotóxicos consiste em primeiro lugar em minimizar os factores de risco cardiovascular, idealmente antes de iniciar uma (quimio)terapia (quimiotóxica) potencialmente cardiotóxica. Por exemplo, pode ser necessário um controlo rigoroso da pressão sanguínea. O trabalho pré-clínico também sugere um papel para os co-estressores. Se a LVEF for limitada, recorre-se à terapia de insuficiência cardíaca usando bloqueadores beta e inibidores da ECA como drogas de primeira linha. Os dados pré-clínicos sugerem que a via beta-adrenérgica cardíaca também actua via MEK e poderia ser favoravelmente influenciada pelo bloqueio. No caso de uma limitação conhecida da LVEF ou de um perfil de alto risco, a terapia profiláctica da insuficiência cardíaca pode ser considerada [19].
A monitorização próxima dos biomarcadores cardíacos (troponina e NT-BNP), bem como a monitorização regular do ECG (especialmente prolongamento do QT, arritmias) durante a terapia pode fornecer indicações precoces de uma reacção cardiotóxica. Além disso, os sintomas clínicos de insuficiência cardíaca, tais como edema ou dispneia, devem ser avaliados regularmente. Um exame cardiológico, incluindo ecocardiografia, deve ser realizado a cada 12 semanas durante a terapia. Para além da FEVE sistólica, podem tornar-se aparentes reduções precoces da função diastólica ou longitudinal ou aumentos da pressão arterial pulmonar. Com base em relatórios de casos, pode assumir-se uma ocorrência precoce de disfunção cardíaca para os inibidores MEK nos primeiros 14 dias após o início da terapia. O controlo anterior com ecocardiografia e marcadores cardíacos parece, portanto, razoável. Um possível esquema para o tratamento de doentes de alto risco devido a factores terapêuticos ou predisponentes pode ser encontrado na figura 2.
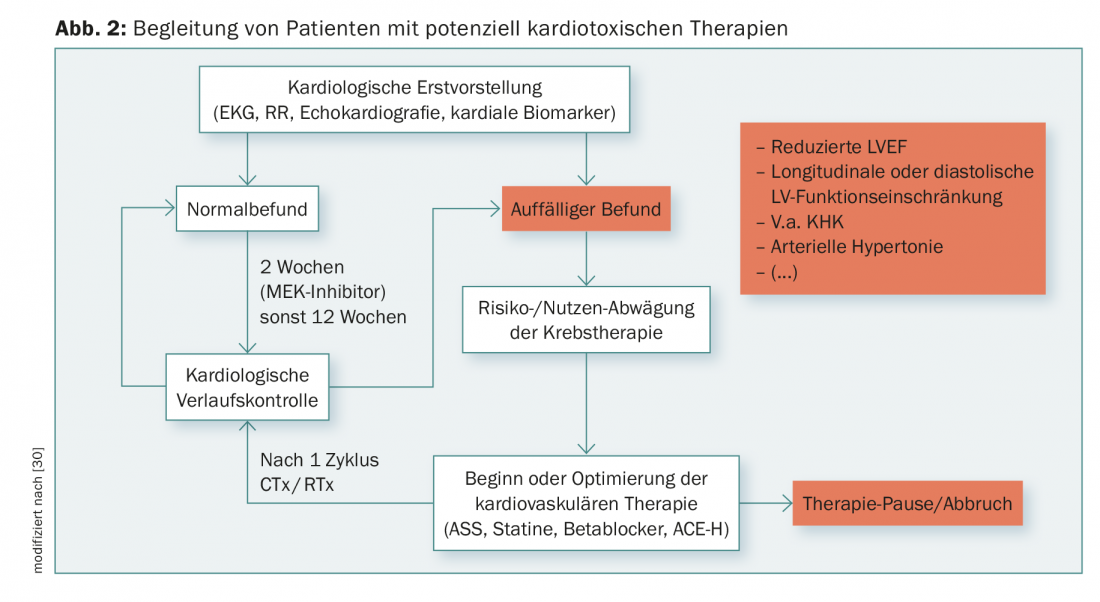
No futuro, os alvos específicos das kinases MAP e vias de sinalização associadas podem ser identificados com base no conhecimento molecular, o que tornará possível um tratamento eficiente dos efeitos secundários cardíacos.
Mensagens Take-Home
- Os efeitos secundários cardíacos da inibição de BRAF/MEK têm estado, até agora, na faixa percentual de um dígito. No entanto, quando ocorrem, podem levar a graves consequências e a uma interrupção da terapia.
- Segundo o CES, o tratamento dos efeitos cardiotóxicos consiste em primeiro lugar na minimização dos factores de risco cardiovascular – idealmente antes de se iniciar uma terapia potencialmente cardiotóxica.
- Depois disso, a monitorização próxima dos biomarcadores cardíacos, bem como a monitorização e avaliação regular dos sintomas clínicos por ECG durante a terapia podem fornecer indicações precoces de uma reacção cardiotóxica.
- Uma abordagem interdisciplinar por parte dos oncologistas e cardiologistas do tratamento melhora o prognóstico cardíaco individual.
Literatura:
- Dummer R, et al: As directrizes suíças actualizadas de 2016 para o tratamento e acompanhamento do melanoma cutâneo. Swiss Med Wkly 2016; 146: w14279.
- Cheng L, et al: Testes moleculares para mutações BRAF para informar as decisões de tratamento do melanoma: um movimento em direcção à medicina de precisão. Mod Pathol 2018; 31(1): 24-38.
- Long GV, et al: Dabrafenib e trametinib versus dabrafenib e placebo para o melanoma Val600 BRAF-mutant: um ensaio controlado multicêntrico, duplo-cego, fase 3 aleatorizado. Lancet 2015; 386(9992): 444-451.
- Robert C, et al: Melhoria da sobrevivência global no melanoma com dabrafenibe e trametinibe combinados. N Engl J Med 2015; 372(1): 30-39.
- Chapman PB, et al: Melhoria da sobrevivência com vemurafenibe em melanoma com mutação de BRAF V600E. N Engl J Med 2011; 364(26): 2507-2516.
- Hauschild A, et al: Dabrafenib em melanoma metastático mutado BRAF: um ensaio controlado multicêntrico, com rótulo aberto, fase 3 aleatorizado. Lancet 2012; 380(9839): 358-365.
- Long GV, et al: Inibição combinada de BRAF e MEK versus inibição de BRAF apenas no melanoma. N Engl J Med 2014; 371(20): 1877-1888.
- Larkin J, et al: Vemurafenibe e cobimetinibe combinados em melanoma mutilado por BRAF. N Engl J Med 2014; 371(20): 1867-1876.
- Flaherty KT, et al: Melhoria da sobrevivência com inibição de MEK no melanoma mutilado por BRAF. N Engl J Med 2012; 367(2): 107-114.
- Ribas A, et al: BRIM-2: Um estudo aberto, multicêntrico fase II do vemurafenibe em doentes previamente tratados com melanoma metastático positivo de mutação BRAF V600E. Journal of Clinical Oncology 2011; 29(15 suppl): 8509-8509.
- Flaherty L, et al: Um estudo de acesso alargado de vemurafenibe em doentes com melanoma metastático nos Estados Unidos, com um único braço, rótulo aberto. Cancro J 2014; 20(1): 18-24.
- Larkin J, et al: Vemurafenib em doentes com BRAF(V600) melanoma metastático mutante: um estudo de segurança de rótulo aberto, multicêntrico. Lancet Oncol 2014; 15(4): 436-444.
- Wang Y: Quinases proteicas activadas por mitogen no desenvolvimento e doenças do coração. Circulação 2007; 116(12): 1413-1423.
- Molkentin JD: Parsing good versus bad signalling paths in the heart: role of calcineurin-nuclear factor of activated T-cells. Circ Res 2013; 113(1): 16-19.
- Marin TM, et al: Rapamycin inverte a cardiomiopatia hipertrófica num modelo de rato da mutação PTPN11 associada à síndrome LEOPARD. J Clin Invest 2011; 121(3): 1026-1043.
- Wu X, et al: MEK-ERK modulation pathway modulation ameliorates disease phenotypes in a mouse model of Noonan syndrome associated with the Raf1(L613V) mutation. J Clin Invest 2011; 121(3): 1009-1025.
- Bueno OF, et al: O caminho de sinalização MEK1-ERK1/2 promove a hipertrofia cardíaca compensada em ratos transgénicos. EMBO J 2000; 19(23): 6341-6350.
- Harris IS, et al: Raf-1 kinase é necessário para hipertrofia cardíaca e sobrevivência cardiomiocitária em resposta à sobrecarga de pressão. Circulação 2004; 110(6): 718-723.
- Zamorano JL, et al.: 2016 ESC Position Paper on cancer treatments and cardiovascular toxicity developed under the auspices of the ESC Committee for Practice Guidelines: The Task Force for cancer treatments and cardiovascular toxicity of the European Society of Cardiology (ESC). Eur Heart J 2016; 37(36): 2768-2801.
- Krenz M, Yutzey KE, Robbins J: A mutação da síndrome do Noonan Q79R em Shp2 aumenta a proliferação de células mesenquimais de primórdios valvares através de sinalização kinase 1/2 extracelular regulada por sinal. Circ Res 2005; 97(8): 813-820.
- Carta C, et al: As mutações do missense Germline que afectam a isoforma B da KRAS estão associadas a um fenótipo grave da síndrome de Noonan. Am J Hum Genet 2006; 79(1): 129-135.
- Niihori T, et al: Mutações de Germline KRAS e BRAF na síndrome cárdio-fácio-cutânea. Nat Genet 2006; 38(3): 294-296.
- Aoki Y, et al.: Mutações da linha germinal na SARH proto-oncogene causam a síndrome de Costello. Nat Genet 2005; 37(10): 1038-1040.
- Sheng Z, et al.: Inibição da cardiotrofina 1 (CT-1) da apoptose do miócito cardíaco através de uma via dependente de proteína mitogénica activada por kinase-dependente. Divergência dos sinais a jusante do CT-1 para hipertrofia da célula miocárdica. J Biol Chem 1997; 272(9): 5783-5791.
- Lips DJ, et al: MEK1-ERK2 sinalling pathway protege o miocárdio de lesões isquémicas in vivo. Circulação 2004; 109(16): 1938-1941.
- De Windt LJ, et al: A hipertrofia mediada pela calcineurina protege os cardiomiócitos da apoptose in vitro e in vivo: Um modelo independente da apoptose de insuficiência cardíaca dilatada. Circ Res 2000; 86(3): 255-263.
- Aikawa R, et al.: O stress oxidativo activa quinases extracelulares reguladas por sinal através de Src e Ras em miócitos cardíacos cultivados de ratos neonatais. J Clin Invest 1997; 100(7): 1813-1821.
- Adderley SR, Fitzgerald DJ: Os danos oxidativos dos cardiomiócitos são limitados pela indução extracelular regulada de kinases 1/2-mediada de ciclooxigenase-2. J Biol Chem 1999; 274(8): 5038-5046.
- Ramirez MT, et al: A via MEKK-JNK é estimulada por receptor alfa-1-adrenérgico e activação ras e está associada à hipertrofia cardíaca in vitro e in vivo. J Biol Chem 1997; 272(22): 14057-14061.
- Tilemann LM, et al: Cardio-oncologia: prioridades conflituosas de tratamento anticancerígeno e resultado cardiovascular. Clin Res Cardiol 2018; 107(4): 271-280.
InFo ONCOLOGy & HaEMATOLOGy 2018; 6(4): 8-12.