
As células T dotadas de especificidade tumoral através da expressão de um recetor de antigénio quimérico (CAR) estão a tornar-se cada vez mais importantes. São já cada vez mais utilizadas na terapia celular adotiva na luta contra o cancro. A principal vantagem da transferência de um CAR, por oposição à transferência de um recetor de células T (TCR) normal, é que um CAR pode reconhecer o tumor independentemente do MHC.
As células T dotadas de especificidade tumoral através da expressão de um recetor de antigénio quimérico (CAR) estão a tornar-se cada vez mais importantes. São já cada vez mais utilizadas na terapia celular adotiva na luta contra o cancro. O conceito CAR foi originalmente desenvolvido no final da década de 1980 por Zelig Eshhar (Weizmann Institute of Science, Rehovot, Israel) [1,2]. A maioria dos CAR é constituída por uma construção scFv derivada de anticorpos que se ligam ao antigénio tumoral (fragmento variável de cadeia simples, que é uma proteína de fusão produzida artificialmente, constituída por uma parte variável de uma cadeia leve e uma cadeia pesada de uma imunoglobulina) e a parte intracelular da cadeia CD3ζ, a em dó sustenido está ligado a um ou mais domínios coestimuladores [3]. Esta estrutura semelhante a um bloco de construção permite a ativação de células T específicas de antigénio em resposta ao reconhecimento específico de antigénios na superfície de células malignas, iniciado pela ligação de scFv, e subsequente sinalização através da cadeia CD3ζ e do domínio coestimulador [3]. A co-estimulação ocorre geralmente através de CD28 (superfamília de imunoglobulinas) ou através de 4-1BB (superfamília de receptores de TNF) [3]. No entanto, existem também muitos outros formatos. A principal vantagem da transferência de um CAR, por oposição à transferência de um recetor de células T (TCR) normal, é que um CAR pode reconhecer o tumor independentemente do MHC.
Até agora, esta tecnologia tem sido utilizada para desenvolver CARs que visam vários antigénios de superfície celular em tumores sólidos ou hematológicos. As células CAR-T, específicas para antigénios-alvo como o CD19 nas células B ou o antigénio de maturação das células B (BCMA) nos plasmócitos, conduziram a regressões clínicas impressionantes em leucemias, linfomas ou mielomas em vários estudos clínicos [4–6]. Resultados como estes levaram, entre outras coisas a aprovação do tisagenlecleucel para o tratamento da leucemia linfoblástica aguda de células B (LLA), do axicabtagene-ciloleucel para o tratamento do linfoma não-Hodgkin agressivo de células B Brexucabtagene-Autoleucel para o tratamento do linfoma das células do manto, Lisocabtagene-Maraleucel para o tratamento do linfoma das grandes células B e Idecabtagene-Vicleucel e Ciltacabtagene-Autoleucel para o tratamento do mieloma múltiplo pela Administração de Alimentos e Medicamentos dos EUA (FDA) e a Agência Europeia de Medicamentos (EMA) [3].
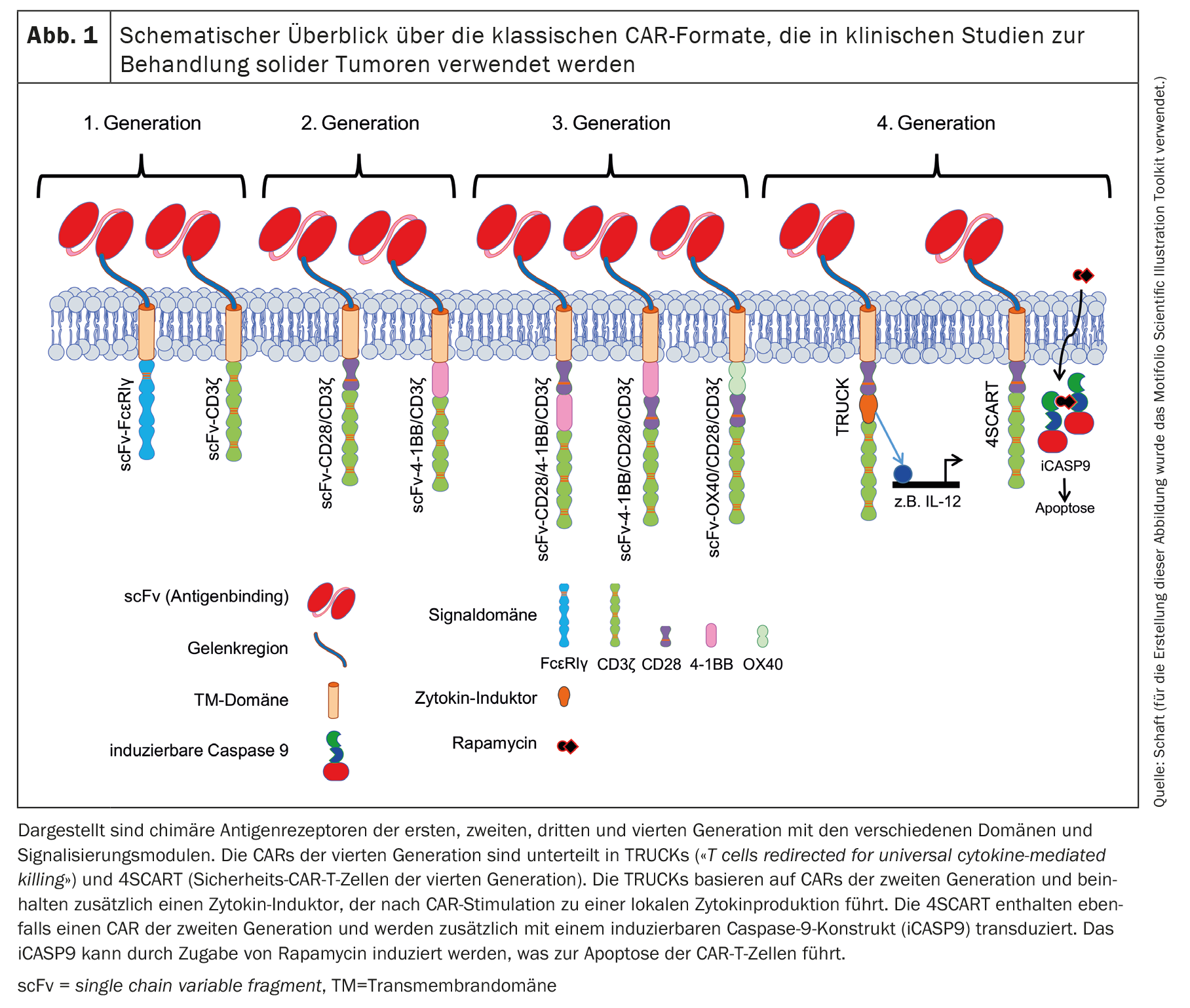
Uma vez que a maioria dos ensaios clínicos se centra na eliminação de tumores hematológicos, o desenvolvimento de células CAR-T contra tumores sólidos está a ficar para trás (ver resumo pormenorizado em [7–11]). Analisando a distribuição geográfica dos ensaios clínicos com células CAR-T contra tumores sólidos registados no Clinicaltrials.gov (n=352; última avaliação em 13 de julho de 2023), é evidente que a maioria destes ensaios está a ser realizada na China (n=199; 55,1%). Os EUA estão em segundo lugar (n=129; 35,7%). Apenas um número muito reduzido de estudos foi efectuado na Europa (Alemanha n=3, Suíça n=1), na Austrália e no resto da Ásia (combinado n=33; 9,2%) (Quadro 1).
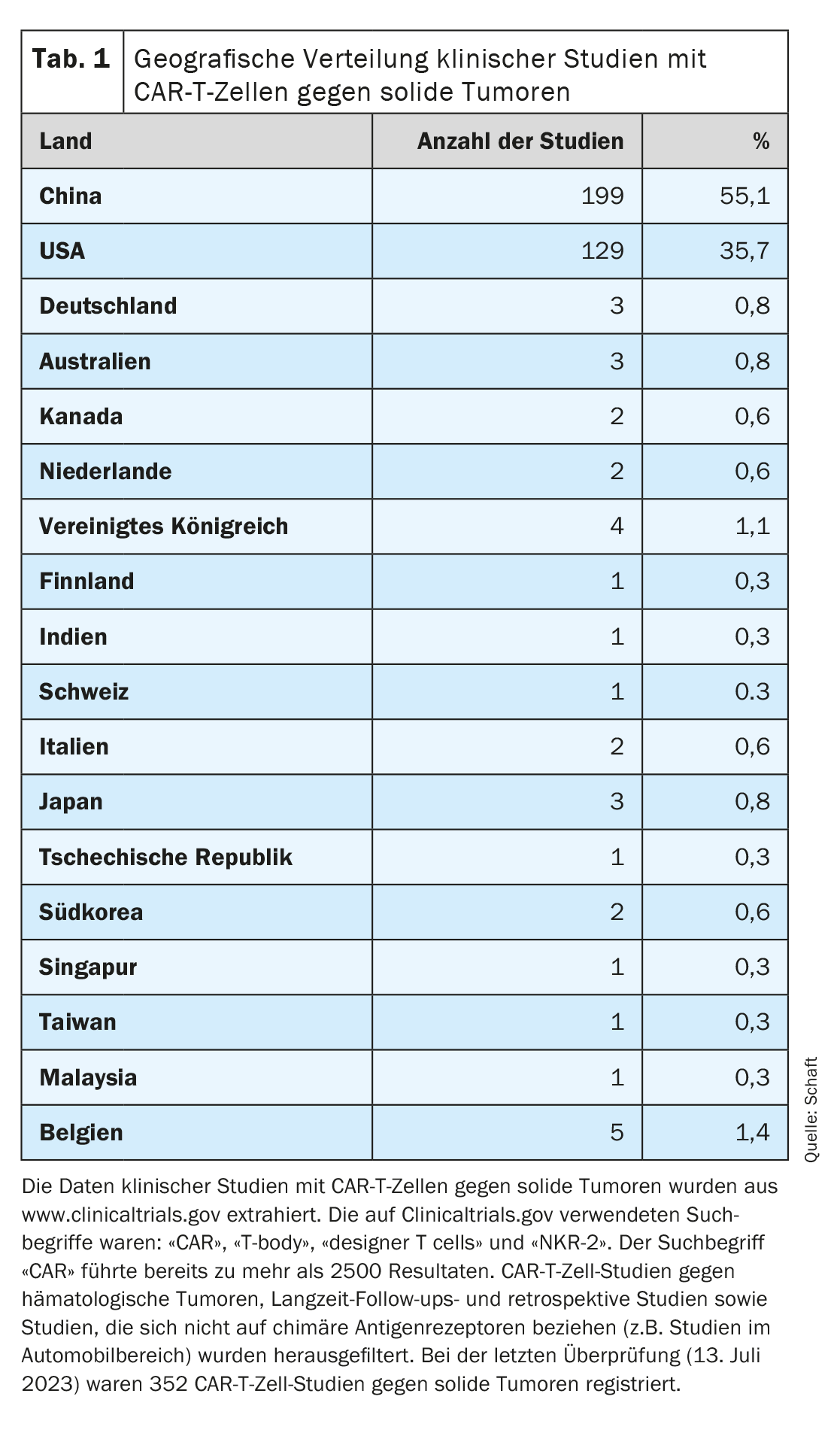
Formatos CAR
Desde a publicação do primeiro conceito de CAR por Zelig Eshhar em 1989 [1,2], os CAR têm sido continuamente desenvolvidos. Isto levou a várias gerações de CAR baseadas no quadro básico do conceito original de CAR. O CAR clássico contém sempre um scFv baseado num anticorpo que pode ligar-se ao antigénio do tumor. Nos CAR de primeira geração (Fig. 1), o scFv está ligado ao domínio de sinalização intracelular do FcεRIγ ou do CD3ζ através de um ligante flexível e de um domínio transmembranar [11,12].
A maioria dos ensaios clínicos registados com células CAR-T contra tumores sólidos utiliza um CAR de segunda geração [11,12], que também contém um domínio coestimulador (Fig. 1). A co-estimulação é geralmente efectuada por CD28 ou 4-1BB [3]. A co-estimulação de CD28 apoia fisiologicamente a produção de IL-2, -6, -10 e outras interleucinas, bem como a progressão do ciclo celular, a sobrevivência, a diferenciação e a função citolítica das células T [13]. Em muitos estudos em que foram utilizados CARs com um domínio de sinalização CD28, observaram-se efeitos antitumorais eficazes e rápidos. No entanto, estes foram apenas de curta duração e foram associados a uma sobrevivência limitada in vivo em comparação com os CAR com um domínio de sinalização 4-1BB, por exemplo [14]. A sinalização 4-1BB fisiológica nas células T aumenta a progressão e a proliferação do ciclo celular, a secreção de citocinas, o potencial citolítico das células T e inibe a eliminação clonal ea morte celular induzida pela ativação (AICD) [15,16]. Os CAR que contêm 4-1BB como domínio de sinalização não só permitiram uma ativação celular mais robusta, aumentaram a persistência in vivo, como também promoveram a diferenciação das células CAR-T emcélulas de memória central [4,14,17–24].
Os CAR de terceira geração [11,12] contêm combinações de domínios coestimuladores: CD28/4-1BB, 4-1BB/CD28 ou OX40/CD28 (Fig. 1) [25,26]. Os veículos automóveis de quarta geração são basicamente veículos automóveis de segunda geração com características adicionais. As TRUCK (células T redireccionadas para a morte universal mediada por citocinas) são modificadas de modo a produzirem citocinas de forma localizada e muito limitada [27]. Os efeitos induzidos dependem do tipo de citocinas libertadas: A IL-12, por exemplo, pode ativar uma resposta imune inata contra o tumor [28], provoca uma redução da suscetibilidade aos efeitos inibitórios das células T reguladoras (Tregs) [29] e aumenta a secreção de citocinas e a proliferação de células T [30,31]. A IL-15, por outro lado, aumenta a atividade antitumoral das células CAR-T [32].
Outra variante da quarta geração é o 4SCART (células CAR-T de segurança). Estas células T são transduzidas simultaneamente com um CAR e uma caspase 9 induzível (iCASP9) como precaução de segurança contra eventos adversos. O iCASP9 pode ser induzido pela adição de rapamicina, o que leva à apoptose das células CAR-T.
Transferir tecnologias
Um requisito essencial na produção de células T CAR é encontrar um método adequado para transferir o CAR para as células T. Para o efeito, podem ser utilizados vários métodos existentes. A maioria dos ensaios clínicos utiliza um método de transferência viral (retroviral ou lentiviral) para introduzir de forma estável o CAR nas células T. Durante este processo, um gene codificador de CAR é transportado do vírus para a célula T, onde é integrado de forma estável no ADN genómico. A descendência destas células transduzidas é portadora do gene CAR e pode expressar o recetor na sua superfície celular. As desvantagens da transdução viral são a integração aleatória no genoma da célula hospedeira, que pode levar à destruição ou ativação de alguns genes (ou seja, mutagénese insercional), bem como a introdução de material/genes virais. Este método pode provocar efeitos secundários graves nos doentes tratados com células CAR-T. Lamers et al. descreveram, por exemplo, o desenvolvimento de respostas imunitárias ao transgene que codifica o recetor e ao vetor retroviral [33].
Alguns ensaios clínicos utilizam um sistema de entrega de genes não viral ou um método de transferência que integra o gene CAR num local específico (por exemplo, sistema de transposão Sleeping Beauty [34–37], sistema de transposão PiggyBac [36,37], CRISPR-Cas9 [38]). A transfecção de ADN ou ARN são outros sistemas de transferência [39], que, no entanto, não conduzem à integração da sequência codificadora CAR no genoma da célula hospedeira. A expressão transitória do CAR daí resultante tem algumas vantagens.
Células CAR-T contra tumores sólidos – seleção de antigénios e precauções de segurança
Tal como acima descrito, a utilização clínica de células CAR-T no tratamento de tumores sólidos está a ficar para trás em relação ao sucesso das células CAR-T no tratamento de tumores hematológicos. Uma das razões para tal é que o CD19 e o BCMA são antigénios alvo que são especificamente expressos por células B ou plasmócitos e a sua eliminação completa é relativamente inofensiva. Outros antigénios, especialmente em tumores sólidos, são frequentemente também expressos em tecidos saudáveis, o que torna difícil a seleção de um antigénio alvo adequado.
Seleção do antigénio
Os antigénios alvo ideais em tumores sólidos combinam três propriedades essenciais:
- Expressão uniforme na superfície das células malignas, o que reduz o risco de variantes de fuga negativas para o antigénio.
- Ausência de expressão em células não malignas (ou seja, expressão exclusiva nas células tumorais), evitando assim o risco de efeitos secundários graves e potencialmente fatais decorrentes da atividade on-target/off-tumour das células CAR-T [40,41].
- Um papel crucial como condutor oncogénico nas células cancerígenas que impede a desregulação dos antigénios devido a uma vantagem selectiva de sobrevivência das células malignas.
- Além disso, a co-expressão do antigénio em células vizinhas no microambiente tumoral (por exemplo, em vasos associados ao tumor, fibroblastos e macrófagos) é outra caraterística positiva de um antigénio alvo ideal, uma vez que a estrutura de fornecimento do tumor pode ser atacada pela terapia específica do antigénio.
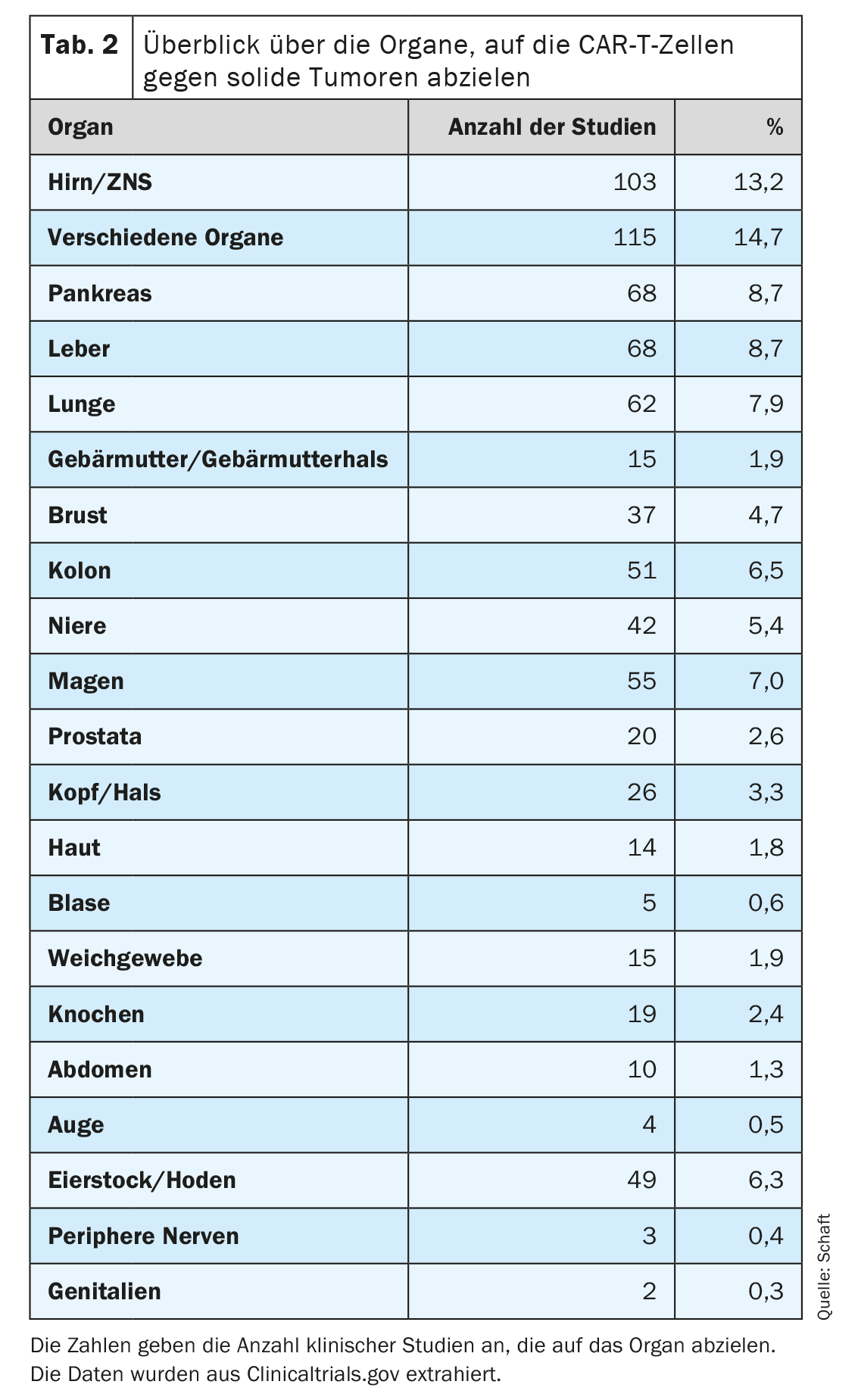
O segundo ponto, em particular, coloca o maior problema para o desenvolvimento de células CAR-T contra tumores sólidos, uma vez que a maioria dos antigénios expressos em tumores sólidos são também expressos em tecidos saudáveis importantes. Isto pode levar a uma reação indesejável no alvo/fora do tumor e a efeitos secundários associados. No entanto, muitos tipos diferentes de tumores sólidos (81 entidades cancerígenas no total) num total de 20 órgãos estão a ser alvo de células CAR-T específicas para 63 antigénios diferentes (Quadro 2). Em muitos estudos clínicos, estão a ser investigados, em particular, tumores do cérebro/CNS, do fígado, do pâncreas e do pulmão (n=103, 68, 68 e 62, respetivamente; Tab. 2). Tal pode dever-se à elevada necessidade médica e/ou à falta de terapias alternativas eficazes para os tumores nos órgãos correspondentes.
Precauções de segurança
Se um antigénio alvo for reconhecido pelas células CAR-T transferidas em tecidos saudáveis, podem ocorrer efeitos secundários indesejáveis e graves. Foram desenvolvidas várias estratégias para desativar as células CAR-T o mais rapidamente possível em caso de toxicidade no doente. A rapamicina, uma molécula capaz de induzir a dimerização dos constructos, pode ser utilizada, por exemplo, para ativar uma caspase 9 induzível. No 4SCART, estas construções induzíveis são transferidas para as células T em simultâneo com o CAR, como o chamado interrutor suicida (Fig. 1). Após a dimerização induzida pela rapamicina, a caspase 9 induz a apoptose das células T CAR. Isto também elimina a atividade indesejada/inesperada das células T contra o tecido saudável(efeitos no alvo/fora do tumor) [42,43]. Outros comutadores possíveis, como a estratégia timidina quinase/ganciclovir do vírus do herpes simplex (HSV-tk/GCV) [44,45] já estão a ser utilizados [11].
Uma medida de segurança especial para contornar a autoimunidade prolongada induzida por uma reação on-target/off-tumoral do CAR é a transfecção do CAR por electroporação do ARNm [11]. Já demonstrámos em várias publicações que a transfecção transiente de células T com CARs utilizando a electroporação de mRNA pode ser uma ferramenta eficaz e segura na imunoterapia do cancro [46-50]. O processo de electroporação baseia-se em mecanismos físico-químicos complexos que conduzem à perfuração da membrana plasmática através da aplicação de campos eléctricos e permitem a subsequente entrada do ARNm no citosol [51]. A utilização de células CAR-T transfectadas com ARN oferece a vantagem de a expressão do recetor ser limitada no tempo, pelo que a potencial toxicidade fora do alvo e no alvo/fora do tumor é também limitada. A estratégia de transferência de CAR-RNA é particularmente atractiva em ensaios clínicos de fase 0/1 que investigam novos antigénios tumorais para a terapia com células T CAR com um perfil de segurança clínica desconhecido.
Células CAR-T clinicamente testadas contra o melanoma uveal
Surpreendentemente, apenas quatro estudos clínicos com células T CAR contra tumores sólidos se centram no olho (Quadro 2) . Destes, dois estudos são dirigidos contra o retinoblastoma e dois estudos contra o melanoma uveal. O melanoma da úvea é o tipo mais comum de cancro do olho e provoca metástases em até 50% dos doentes. As metástases ocorrem predominantemente no fígado e estão associadas a um tempo de sobrevivência mediano fraco de cerca de 12 meses. Apesar dos enormes progressos no tratamento do melanoma cutâneo metastizado com o bloqueio do ponto de controlo imunitário (ICB), este não é eficaz no melanoma uveal. Apenas o recém-aprovado iniciador de células T biespecífico Tebentafusp (um scFv específico de CD3 ligado a um TCR solúvel que reconhece um péptido gp100 apresentado pelo HLA-A2) pode atenuar a progressão e prolongar a sobrevivência global num subconjunto de doentes com melanoma uveal metastático. Os efeitos positivos observados do Tebentafusp são de curta duração, com um tempo de sobrevivência global mediano de 22 meses e uma taxa de sobrevivência a três anos de 24%. Além disso, apenas 50% dos doentes com metástases são elegíveis para esta opção de tratamento devido à restrição do HLA-A2. Por conseguinte, existe também uma grande necessidade médica de abordagens de tratamento alternativas para esta entidade tumoral.
Dois ensaios clínicos com células CAR-T constantes da lista internacional da US National Library of Medicine (www.clinicaltrials.gov) estão atualmente a recrutar doentes com melanoma uveal (NCT03893019 contra o cluster of differentiation 20 [CD20] e NCT03635632 contra o disialogangliosídeo GD2 [GD2]). Ambos os estudos utilizam células CAR-T que visam antigénios não específicos do melanoma.
O primeiro ensaio clínico (fase 1; NCT03893019), que utiliza células CAR-T específicas para CD20 de segunda geração (Fig. 2A), é patrocinado pela Miltenyi Biomedicine GmbH (Investigador Principal [PI]: Peter Borchmann; Hospital Universitário de Colónia) e está a recrutar principalmente doentes com melanoma cutâneo. Alguns doentes com melanoma uveal também são tratados. O CD20 é um antigénio alvo que é expresso em células B normais e é normalmente utilizado como antigénio alvo no linfoma não Hodgkin de células B [52]. No entanto, também é expresso num pequeno subconjunto de células de melanoma [53,54]. No entanto, o tratamento orientado de um antigénio que só é expresso num pequeno subgrupo de células cancerígenas poderia levar a que o tumor escapasse facilmente à terapia com células CAR-T. Infelizmente, o estado atual deste ensaio clínico não é conhecido.
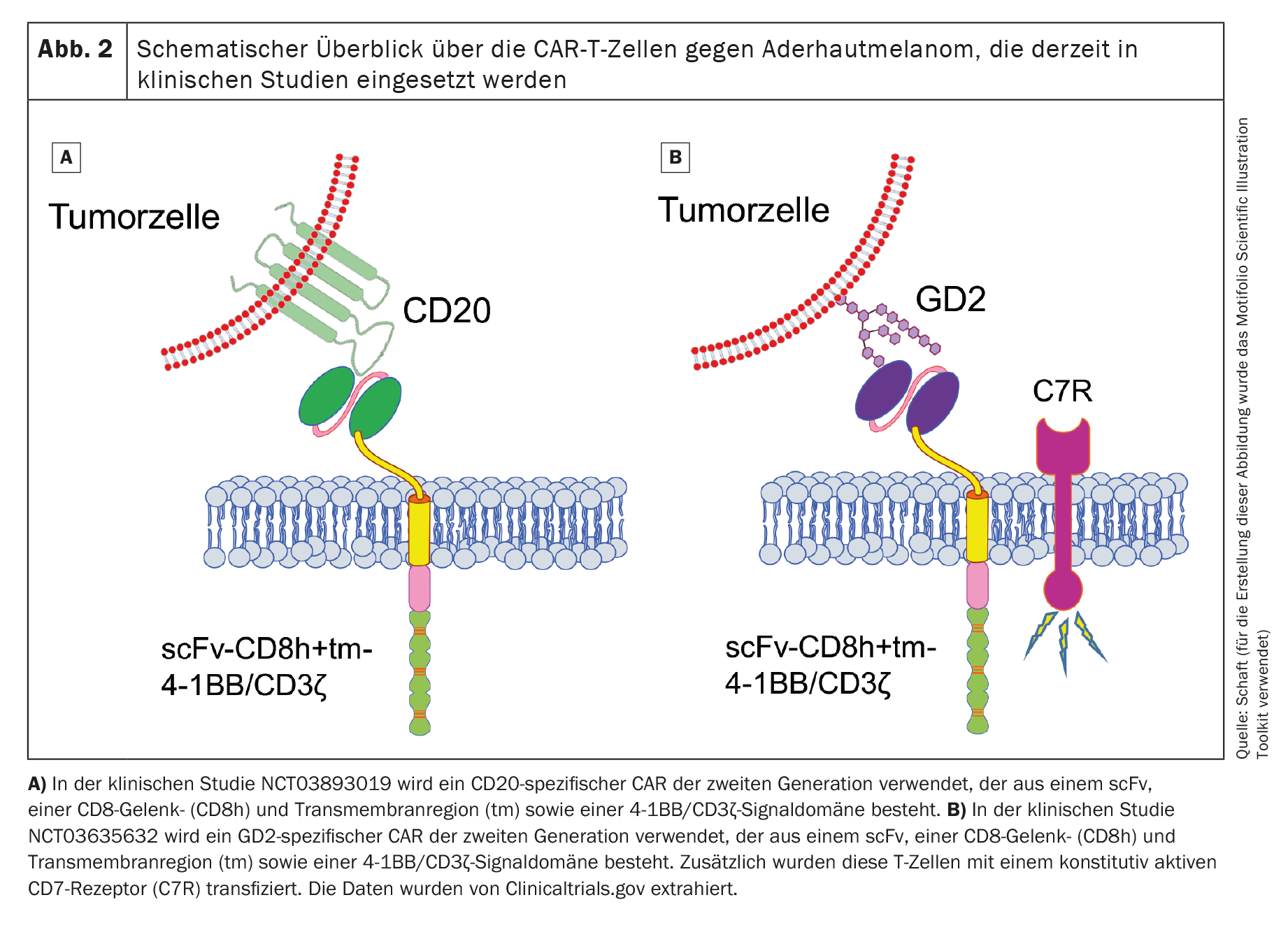
O segundo ensaio clínico (também de fase 1; NCT03635632) utiliza células CAR-T específicas para GD2 (Fig. 2B) e está a recrutar doentes com neuroblastoma, sarcoma, melanoma uveal, cancro da mama ou outros cancros que expressem GD2. Este estudo, patrocinado pelo Baylor College of Medicine (PI: Bilal Omer; Baylor College of Medicine), está atualmente ativo, mas não estão a ser recrutados quaisquer doentes.
Para além do CAR de segunda geração, os investigadores também transduziram um recetor de IL-7 constitutivamente ativo nas células T, a fim de prolongar a sobrevivência das células CAR-T após a transferência adotiva. A GD2 é expressa, embora em níveis muito baixos, no cerebelo e nos nervos periféricos [55], o que torna o tratamento com células CAR-T específicas da GD2 muito arriscado se for induzida uma reação on-target/off-tumor. Também ainda não foram publicados dados relativos a este ensaio clínico.
Em resumo, existe um grande potencial para ensaios clínicos com células CAR-T contra antigénios específicos do melanoma (úvea) em que o risco de uma reação no alvo/fora do tumor é baixo.
A procura de um melhor antigénio tumoral no melanoma uveal
A prevenção ou redução de uma possível reação on-target/off-tumour é, como já foi referido na secção “Células CAR-T contra tumores sólidos”, um requisito básico na procura de novos antigénios. Em termos pré-clínicos, a atenção centra-se atualmente em dois antigénios expressos nos melanomas uveais: o recetor 2 do fator de crescimento epidérmico humano (HER2) e o proteoglicano de sulfato de condroitina 4 (CSPG4).
HER2
O HER2 é um membro da família ErbB de receptores tirosina-quinases (EGFR [ErbB-1], HER2 [/neu] [ErbB-2], Her 3 [ErbB-3] e Her 4 [ErbB-4]). As mutações no HER2 levam a uma sobreexpressão, resultando numa ativação constitutiva e numa divisão celular descontrolada. Isto aplica-se sobretudo ao cancro da mama, mas também a outros tipos de cancro, como o cancro do ovário ou os gliomas [56–58].
Como já foi referido, a utilização de células CAR-T é uma faca de dois gumes, uma vez que a eficácia destas células pode também virar-se contra o doente [59]. Nunca se pode excluir que um tipo de célula raro mas essencial expresse o antigénio em tecidos saudáveis. Investigadores do Instituto Nacional do Cancro relataram um caso que ilustra o potencial mortal da toxicidade on-target/off-tumoral do antigénio HER2. Pouco depois da infusão de células CAR-T específicas de HER2, foram observados sintomas clínicos de síndrome de dificuldade respiratória aguda que exigiram respiração artificial num doente com cancro colorrectal metastático [60]. Infelizmente, o doente morreu cinco dias após o início da angústia respiratória aguda [60]. A causa da morte foi provavelmente o resultado de toxicidade on-target/off-tumoral causada por baixos níveis de HER2 nas células epiteliais dos pulmões. Notavelmente, o CAR foi baseado no anticorpo monoclonal trastuzumab aprovado pela FDA, que tem sido utilizado extensivamente sem causar toxicidade pulmonar grave [61]. Isto sublinha a necessidade de uma seleção muito cuidadosa do antigénio alvo para a terapia com células T CAR.
CSPG4
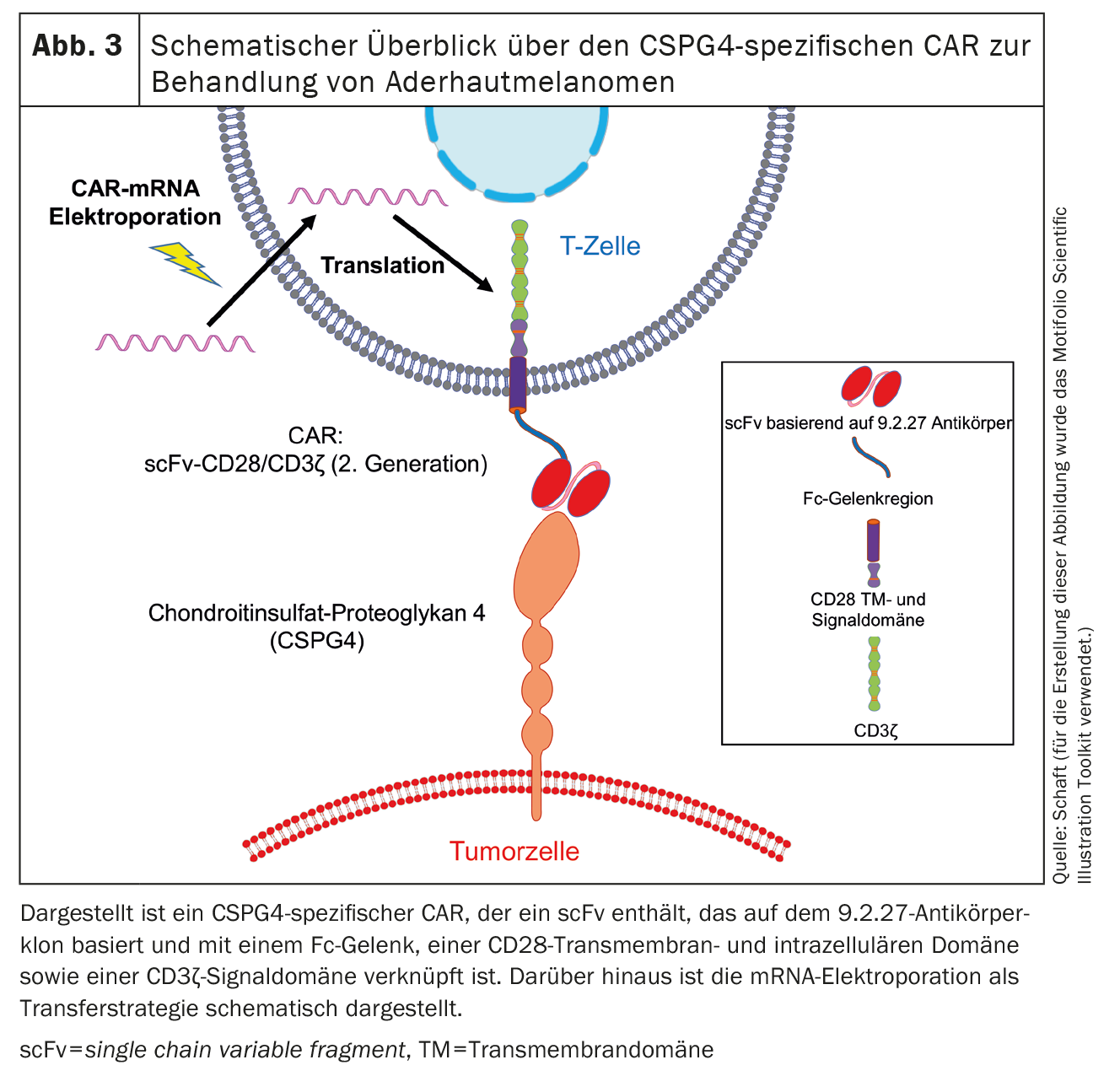
O segundo antigénio expresso nos melanomas uveais é o proteoglicano de sulfato de condroitina 4 (CSPG4) (Fig. 3), anteriormente também conhecido como proteoglicano de sulfato de condroitina associado ao melanoma (MCSP) ou antigénio associado ao melanoma de elevado peso molecular (HMW-MAA). A CSPG4 é uma proteína transmembranar de passagem única do tipo 1 e foi descoberta por Ralph Reisfeld [72]. Nós e outros grupos de trabalho [47-50, 62-71] trabalhámos principalmente no CSPG4. A expressão de CSPG4 está associada a um aumento da proliferação e da sobrevivência das células tumorais. Este processo é iniciado pela ativação da via de sinalização MAPK e pela apresentação cruzada de factores de crescimento [73]. Além disso, a CSPG4 desempenha um papel na motilidade celular e na infiltração de tecidos devido à sua associação com o citoesqueleto de actina e à ligação a várias integrinas e componentes da matriz extracelular [74]. Além disso, a CSPG4 está envolvida na formação da placenta [75], na angiogénese [76], na formação de redes neuronais [77], na renovação dos queratinócitos e na homeostasia das células estaminais epidérmicas [78].
Várias publicações descreveram a expressão de CSPG4 em tecidos não patológicos, tais como precursores do folículo piloso e células epidérmicas, células endoteliais e pericitos activados (mas não em vasos maduros) [79,80], condrócitos da cartilagem articular [81], células musculares lisas [82] e células da sinapse neuromuscular do músculo esquelético humano pós-natal [83]. No entanto, a expressão de CSPG4 é significativamente menor nos tecidos saudáveis do que nas células tumorais [62,73,84].
Beard et al. mostraram que a CSPG4 podia ser detectada ao nível do ARN numa variedade de tecidos normais, incluindo o sistema nervoso central, o olho, a pele, o tecido adiposo, os vasos sanguíneos, a bexiga, o trato gastrointestinal, o útero, a próstata, o baço e o timo [85]. Em média, o ARN do CSPG4 está sobreexpresso 6,6 vezes no tecido maligno (melanoma) em comparação com o tecido saudável [85]. Estes resultados confirmam trabalhos anteriores de Erfurt et al. que demonstraram que, embora o ARNm da CSPG4 tenha sido detectado em algumas amostras de tecido normal, a expressão estava significativamente aumentada em amostras de melanoma cutâneo e melanoma uveal [86].
A coloração imuno-histoquímica e as matrizes proteicas de fase reversa mostraram que a expressão específica de CSPG4 a nível proteico só podia ser detectada em algumas amostras do intestino delgado [63]. Não foi detectada a expressão da proteína CSPG4 nos seguintes tecidos: Cérebro, nervos periféricos, pele, mesotélio, mama, coração, rim, glândulas supra-renais, fígado, pulmão, gânglios linfáticos, músculos, ovários, pâncreas, esófago, próstata, baço, estômago, útero e tiroide [62,63]. Em contrapartida, a CSPG4 é expressa em quase todas as células de melanoma cutâneo [87–90]. Os melanomas coroidais [91,92] e alguns outros tumores como sarcomas, astrocitomas, gliomas, neuroblastomas [93-96], leucemias [97–101] e cancro da mama triplo-negativo também expressam CSPG4 [102]. Em muitas destas doenças malignas, a expressão de CSPG4 está associada a um mau prognóstico e a um crescimento agressivo do tumor [103].
Além disso, o CSPG4 é considerado um antigénio alvo primário dos tumores [84], uma vez que desempenha um papel na metástase do melanoma [104] e é expresso em pericitos activados durante a angiogénese nos tumores e na hipoxia [105–107]. Este último permite a seleção da vasculatura do tumor. Mais importante ainda, a CSPG4 actua como um motor oncogénico no melanoma e promove o crescimento e a sobrevivência das células malignas após a ativação de várias vias de sinalização [73]. Por conseguinte, o tumor não pode simplesmente reduzir a regulação da CSPG4 para escapar à terapia direccionada para a CSPG4.
Por este motivo, o CSPG4 já foi selecionado como antigénio alvo por vários grupos e os CAR específicos do CSPG4 foram introduzidos nas células T através de vários mecanismos. CARs específicos de CSPG4 de diferentes formatos, que foram transduzidos de forma viral em células T, resultaram numa forte citotoxicidade das células T in vitro . Em modelos animais, as células T transferidas adotivamente reagiram a vários tumores que expressam CSPG4, como o melanoma, o cancro da mama, o mesotelioma, o glioblastoma e o osteossarcoma [47–50,62–71]. Geldres et al. transduziram retroviralmente um CAR específico para CSPG4 de segunda geração em células T. In vitro, estas células T CAR específicas de CSPG4 foram capazes de reconhecer e lisar células de melanoma de uma forma específica para o antigénio [65]. Além disso, a ligação do antigénio ao CAR levou a uma secreção pronunciada de IL-2 e IFNγ. In vivo , a transferência de células CAR-T específicas de CSPG4 para ratinhos portadores de células de melanoma levou a um abrandamento significativo do crescimento do tumor e a uma melhoria da sobrevivência global dos ratinhos [65]. Na mesma publicação, foi descrita a ausência de reatividade positiva das células CAR-T específicas de CSPG4 a tecido normal com ARN de CSPG4 detetável mas sem expressão da proteína CSPG4 [65]. Conseguiram demonstrar que as células CAR-T específicas de CSPG4 não exerciam uma citotoxicidade significativa contra linhas celulares epiteliais primárias da próstata, do pulmão e do rim [65]. Por conseguinte, as análises de expressão efectuadas por Beard et al. [63,85] atenuam as preocupações acima descritas relativamente àtoxicidade no alvo/fora do tumor induzida pelas células CAR-T específicas de CSPG4, uma vez que a expressão de CSPG4 a nível proteico é necessária para induzir uma reatividade indesejada das células CAR-T.
Até agora, utilizámos o método de transfecção de ARNm por electroporação para introduzir CARs nas células T. Neste contexto, já testámos vários CAR específicos para a CSPG4 e observámos que as células CAR-T transfectadas com mRNA são capazes de eliminar as células tumorais de uma forma específica para o antigénio. A cinética de expressão dos CAR transferidos por electroporação depende da espinha dorsal do CAR [47]. Foi identificado um CAR que apresenta uma elevada expressão nas células T e uma elevada reatividade anti-células tumorais. Este CAR contém um scFv baseado no clone de anticorpo 9.2.27 ligado a um espaçador Fc, um domínio transmembranar e intracelular CD28 e um domínio de sinalização CD3ζ [47](Fig. 3). Experiências in vivo com ratinhos imunodeficientes Rag-/-/common γ-chain-/- mostraram que as células CAR-T específicas de CSPG4 transfectadas prolongaram significativamente o tempo médio de sobrevivência dos ratinhos [47].
A fim de transferir as células CAR-T específicas de CSPG4 para a aplicação clínica, a produção de células CAR-T à escala clínica foi estabelecida em laboratório por transfecção de mRNA de um CAR em total conformidade com as BPF [50]. Isto demonstrou que é possível a produção repetida de um número suficiente de células T altamente puras, específicas de CSPG4, transfectadas com CAR. Estas células T modificadas apresentam uma eficiência de transfecção muito elevada, uma elevada expressão de CAR e uma elevada eficácia na destruição das células alvo do melanoma [50].
Embora o CSPG4 seja um antigénio alvo tumoral primário, particularmente no melanoma cutâneo, também descobrimos que não era expresso em várias linhas celulares de melanoma uveal que testámos. Por conseguinte, estabelecemos uma plataforma combinada plataforma para identificar novos antigénios de superfície celular específicos de tumores de melanomas uveais. Utilizando esta plataforma, identificámos uma proteína candidata como um antigénio alvo adequado no melanoma uveal para o desenvolvimento de outros CAR. Os CARs gerados que podem ligar-se a esta proteína candidata estão atualmente a ser testados quanto à sua funcionalidade e especificidade.
Perspectivas
A utilização de células CAR-T no tratamento de tumores sólidos tem um grande potencial. São necessários mais estudos pré-clínicos e ensaios clínicos para responder à elevada necessidade médica de tratamento de entidades cancerígenas sólidas (como o melanoma uveal).
Os futuros ensaios clínicos devem centrar-se no teste de novos formatos de CAR. Para além de testar novos domínios extracelulares de ligação a antigénios e novos domínios de sinalização intracelular [109], isto inclui também testar formatos que aumentem a segurança da utilização das células CAR-T [108]. Além disso, novos veículos celulares para transferência de CAR [109,110] prometem alargar o leque de aplicações. Por exemplo, a possibilidade de utilizar células CAR-NK [111] ou células CAR-T alogénicas [112] como padrão pode reduzir os custos da terapia com células CAR e, assim, tornar a terapia acessível a mais doentes.
Além disso, devem ser encontrados mais antigénios específicos do tumor, a fim de evitar reacções no alvo/fora do tumor. Os antigénios que são expressos no estroma tumoral e que podem ser aí atacados pelas células CAR-T são muito promissores nesta área [113]. A orientação de múltiplos antigénios por uma célula CAR-T (ou seja, a expressão de diferentes CAR específicos para diferentes antigénios numa única célula) pode aumentar a especificidade do tumor e reduzir o risco de efeitos fora do alvo. Isto também se aplica ao modelo em que os módulos de sinalização intracelular são divididos entre os diferentes CAR, a fim de aumentar o perfil de segurança das células CAR-T. Isto também torna menos provável o desenvolvimento de variantes de perda de antigénio dos tumores.
Além disso, já estão a ser testadas clinicamente em muitos tumores hematológicos terapias combinadas de células CAR-T com várias pequenas moléculas ou anticorpos monoclonais para impedir os mecanismos de escape dos tumores e aumentar a atividade antitumoral (ver síntese pormenorizada em [114,115]). Estas combinações são igualmente prometedoras para o tratamento de tumores sólidos e devem ser testadas em ensaios clínicos num futuro próximo.
N.S. realizou a pesquisa no clinicaltrials.gov, reviu as figuras e redigiu o manuscrito. S.H. editou o manuscrito. N.S. e S.H. redigiram conjuntamente as perguntas da formação CME.
Literatura:
- Gross G, Gorochov G, Waks T, Eshhar Z: Generation of effector T cells expressing chimeric T cell receptor with antibody type-specificity. Transplant Proc 1989; 21(1 Pt 1): 127–130.
- Gross G, Waks T, Eshhar Z: Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci U S A. 1989; 86(24): 10024–10028.
- June CH, Sadelain M: Chimeric Antigen Receptor Therapy. N Engl J Med 2018; 379(1): 64–73.
- Maude SL, Laetsch TW, Buechner J, et al.: Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N Engl J Med 2018; 378(5): 439-448.
- Neelapu SS, Locke FL, Go WY: CAR T-Cell Therapy in Large B-Cell Lymphoma. N Engl J Med 2018; 378(11): 1065.
- Schuster SJ, Bishop MR, Tam CS, et al.: Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N Engl J Med 2019; 380(1): 45–56.
- Wang Z, Guo Y, Han W: Current status and perspectives of chimeric antigen receptor modified T cells for cancer treatment. Protein Cell 2017; 8(12): 896–925.
- Han S, Latchoumanin O, Wu G, et al.: Recent clinical trials utilizing chimeric antigen receptor T cells therapies against solid tumors. Cancer Lett 2017; 390: 188–200.
- Yeku O, Li X, Brentjens RJ: Adoptive T-Cell Therapy for Solid Tumors. Am Soc Clin Oncol Educ Book 2017; 37: 193–204.
- Arabi F, Torabi-Rahvar M, Shariati A, et al.: Antigenic targets of CAR T Cell Therapy. A retrospective view on clinical trials. Exp Cell Res 2018; 369(1): 1–10.
- Schaft N: The Landscape of CAR-T Cell Clinical Trials against Solid Tumors-A Comprehensive Overview. Cancers (Basel) 2020; 12(9).
- Holzinger A, Abken H: CAR T Cells: A Snapshot on the Growing Options to Design a CAR. Hemasphere 2019; 3(1): e172.
- Boomer JS, Green JM: An enigmatic tail of CD28 signaling. Cold Spring Harb Perspect Biol 2010; 2(8): a002436.
- Kawalekar OU, O’Connor RS, Fraietta JA, et al.: Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity 2016; 44(2): 380–390.
- Cannons JL, Choi Y, Watts TH: Role of TNF receptor-associated factor 2 and p38 mitogen-activated protein kinase activation during 4-1BB-dependent immune response. J Immunol 2000; 165(11): 6193–6204.
- Lee HW, Nam KO, Park SJ, Kwon BS: 4-1BB enhances CD8+ T cell expansion by regulating cell cycle progression through changes in expression of cyclins D and E and cyclin-dependent kinase inhibitor p27kip1. Eur J Immunol 2003; 33(8): 2133–2141.
- Zhao Z, Condomines M, van der Stegen SJC, et al.: Structural Design of Engineered Costimulation Determines Tumor Rejection Kinetics and Persistence of CAR T Cells. Cancer Cell 2015; 28(4): 415–428.
- Milone MC, Fish JD, Carpenito C, et al.: Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol Ther 2009; 17(8): 1453–1464.
- Lim WA, June CH.: The Principles of Engineering Immune Cells to Treat Cancer. Cell 2017; 168(4): 724-740.
- Roselli E, Frieling JS, Thorner K, et al.: CAR-T Engineering: Optimizing Signal Transduction and Effector Mechanisms. BioDrugs 2019; 33(6): 647–659.
- Hombach AA, Holzinger A, Abken H.: The weal and woe of costimulation in the adoptive therapy of cancer with chimeric antigen receptor (CAR)-redirected T cells. Curr Mol Med 2013; 13(7): 1079–1088.
- Sadelain M, Brentjens R, Riviere I: The basic principles of chimeric antigen receptor design. Cancer Discov 2013; 3(4): 388–398.
- Redeker A, Arens R: Improving Adoptive T Cell Therapy: The Particular Role of T Cell Costimulation, Cytokines, and Post-Transfer Vaccination. Front Immunol 2016; 7: 345.
- Weinkove R, George P, Dasyam N, McLellan AD.: Selecting costimulatory domains for chimeric antigen receptors: functional and clinical considerations. Clin Transl Immunology 2019; 8(5): e1049.
- Ceppi F, Rivers J, Annesley C, et al.: Lymphocyte apheresis for chimeric antigen receptor T-cell manufacturing in children and young adults with leukemia and neuroblastoma. Transfusion 2018; 58(6): 1414–1420.
- Li W, Guo L, Rathi P, et al.: Redirecting T Cells to Glypican-3 with 4-1BB Zeta Chimeric Antigen Receptors Results in Th1 Polarization and Potent Antitumor Activity. Hum Gene Ther 2017; 28(5): 437–448.
- Chmielewski M, Hombach AA, Abken H: Of CARs and TRUCKs: chimeric antigen receptor (CAR) T cells engineered with an inducible cytokine to modulate the tumor stroma. Immunol Rev 2014; 257(1): 83–90.
- Chmielewski M, Kopecky C, Hombach AA, Abken H.: IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively Muster an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res 2011; 71(17): 5697–5706.
- Pegram HJ, Lee JC, Hayman EG, et al.: Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood 2012; 119(18): 4133-4141.
- Koneru M, Purdon TJ, Spriggs D, et al.: IL-12 secreting tumor-targeted chimeric antigen receptor T cells eradicate ovarian tumors in vivo. Oncoimmunology 2015; 4(3): e994446.
- Koneru M, O›Cearbhaill R, Pendharkar S, Spriggs DR, Brentjens RJ.: A phase I clinical trial of adoptive T cell therapy using IL-12 secreting MUC-16(ecto) directed chimeric antigen receptors for recurrent ovarian cancer. J Transl Med 2015; 13: 102.
- Xu A, Bhanumathy KK, Wu J, et al.: IL-15 signaling promotes adoptive effector T-cell survival and memory formation in irradiation-induced lymphopenia. Cell Biosci 2016; 6: 30.
- Lamers CH, Willemsen R, van Elzakker P, et al.: Immune responses to transgene and retroviral vector in patients treated with ex vivo-engineered T cells. Blood 2011; 117(1): 72–82.
- Magnani CF, Tettamanti S, Alberti G, et al.: Transposon-Based CAR T Cells in Acute Leukemias: Where are We Going? Cells 2020; 9(6).
- Hudecek M, Ivics Z: Non-viral therapeutic cell engineering with the Sleeping Beauty transposon system. Curr Opin Genet Dev 2018; 52: 100–108.
- Tipanee J, VandenDriessche T, Chuah MK.: Transposons: Moving Forward from Preclinical Studies to Clinical Trials. Hum Gene Ther 2017; 28(11): 1087–1104.
- Vargas JE, Chicaybam L, Stein RT, et al.: Retroviral vectors and transposons for stable gene therapy: advances, current challenges and perspectives. J Transl Med 2016; 14(1): 288.
- Ran FA, Hsu PD, Wright J, et al.: Genome engineering using the CRISPR-Cas9 system. Nat Protoc 2013; 8(11): 2281–2308.
- Birkholz K, Hombach A, Krug C, et al.: Transfer of mRNA encoding recombinant immunoreceptors reprograms CD4+ and CD8+ T cells for use in the adoptive immunotherapy of cancer. Gene Ther 2009; 16(5): 596–604.
- Lamers CH, Sleijfer S, et al.: Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: clinical evaluation and management of on-target toxicity. Mol Ther 2013; 21(4): 904–912.
- Morgan RA, Yang JC, Kitano M, et al.: Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther 2010; 18(4): 843-851.
- Stavrou M, Philip B, Traynor-White C, et al.: A Rapamycin-Activated Caspase 9-Based Suicide Gene. Mol Ther 2018; 26(5): 1266–1276.
- Di Stasi A, Tey SK, Dotti G, et al.: Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med 2011; 365(18): 1673–1683.
- Moolten FL: Tumor chemosensitivity conferred by inserted herpes thymidine kinase genes: paradigm for a prospective cancer control strategy. Cancer Res 1986; 46(10): 5276–5281.
- Beltinger C, Fulda S, Kammertoens T, et al.: Herpes simplex virus thymidine kinase/ganciclovir-induced apoptosis involves ligand-independent death receptor aggregation and activation of caspases. Proc Natl Acad Sci U S A 1999; 96(15): 8699–8704.
- Harrer DC, Simon B, Fujii SI, et al.: RNA-transfection of gamma/delta T cells with a chimeric antigen receptor or an alpha/beta T-cell receptor: a safer alternative to genetically engineered alpha/beta T cells for the immunotherapy of melanoma. BMC Cancer 2017; 17(1): 551.
- Krug C, Birkholz K, Paulus A, et al.: Stability and activity of MCSP-specific chimeric antigen receptors (CARs) depend on the scFv antigen-binding domain and the protein backbone. Cancer Immunol Immunother 2015; 64(12): 1623–1635.
- Dörrie J, Babalija L, Hoyer S, et al.: BRAF and MEK Inhibitors Influence the Function of Reprogrammed T Cells: Consequences for Adoptive T-Cell Therapy. Int J Mol Sci 2018; 19(1).
- Harrer DC, Schuler G, Dörrie J, Schaft N: CSPG4-Specific CAR T Cells for High-Risk Childhood B Cell Precursor Leukemia. Int J Mol Sci 2019; 20(11).
- Wiesinger M, Marz J, Kummer M, et al.: Clinical-Scale Production of CAR-T Cells for the Treatment of Melanoma Patients by mRNA Transfection of a CSPG4-Specific CAR under Full GMP Compliance. Cancers (Basel) 2019; 11(8).
- Shi J, Ma Y, Zhu J, et al.: A Review on Electroporation-Based Intracellular Delivery. Molecules 2018; 23(11).
- Ernst M, Oeser A, Besiroglu B, et al.: Chimeric antigen receptor (CAR) T-cell therapy for people with relapsed or refractory diffuse large B-cell lymphoma. Cochrane Database Syst Rev 2021; 9(9): Cd013365.
- Pinc A, Somasundaram R, Wagner C, et al.: Targeting CD20 in melanoma patients at high risk of disease recurrence. Mol Ther 2012; 20(5): 1056–1062.
- Schmidt P, Kopecky C, Hombach A, et al.: Eradication of melanomas by targeted elimination of a minor subset of tumor cells. Proc Natl Acad Sci U S A 2011; 108(6): 2474–2479.
- Brignole C, Marimpietri D, Pagnan G, et al.: Neuroblastoma targeting by c-myb-selective antisense oligonucleotides entrapped in anti-GD2 immunoliposome: immune cell-mediated anti-tumor activities. Cancer Lett 2005; 228(1–2): 181–186.
- Mitri Z, Constantine T, O’Regan R: The HER2 Receptor in Breast Cancer: Pathophysiology, Clinical Use, and New Advances in Therapy. Chemother Res Pract 2012; 2012: 743193.
- Slamon DJ, Godolphin W, Jones LA, et al.: Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science 1989; 244(4905): 707–712.
- Zhang JG, Kruse CA, Driggers L, et al.: Tumor antigen precursor protein profiles of adult and pediatric brain tumors identify potential targets for immunotherapy. J Neurooncol 2008; 88(1): 65–76.
- Casucci M, Hawkins RE, Dotti G, Bondanza A: Overcoming the toxicity hurdles of genetically targeted T cells. Cancer Immunol Immunother 2015; 64(1): 123–130.
- Morgan RA, Yang JC, Kitano M, et al.: Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther 2010; 18(4): 843–851.
- Slamon DJ, Leyland-Jones B, Shak S, et al.: Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med 2001; 344(11): 783–792.
- Wang Y, Geldres C, Ferrone S, Dotti G: Chondroitin sulfate proteoglycan 4 as a target for chimeric antigen receptor-based T-cell immunotherapy of solid tumors. Expert Opin Ther Targets 2015; 19(10): 1339–1350.
- Beard RE, Zheng Z, Lagisetty KH, et al.: Multiple chimeric antigen receptors successfully target chondroitin sulfate proteoglycan 4 in several different cancer histologies and cancer stem cells. J Immunother Cancer 2014; 2: 25.
- Pellegatta S, Savoldo B, Di IN, et al.: Constitutive and TNFalpha-inducible expression of chondroitin sulfate proteoglycan 4 in glioblastoma and neurospheres: Implications for CAR-T cell therapy. Sci Transl Med 2018; 10(430).
- Geldres C, Savoldo B, Hoyos V, et al.: T lymphocytes redirected against the chondroitin sulfate proteoglycan-4 control the growth of multiple solid tumors both in vitro and in vivo. Clin Cancer Res 2014; 20(4): 962–971.
- Abken H, Hombach A, Heuser C, Reinhold U.: A novel strategy in the elimination of disseminated melanoma cells: chimeric receptors endow T cells with tumor specificity. Recent Results Cancer Res 2001; 158: 249–264.
- Burns WR, Zhao Y, Frankel TL, et al.: A high molecular weight melanoma-associated antigen-specific chimeric antigen receptor redirects lymphocytes to target human melanomas. Cancer Res 2010; 70(8): 3027–3033.
- Losch FO, Muller R, Mutschler B, et al.: Activation of T cells via tumor antigen specific chimeric receptors: the role of the intracellular signaling domain. Int J Cancer 2003; 103(3): 399–407.
- Reinhold U, Liu L, Ludtke-Handjery HC, et al.: Specific lysis of melanoma cells by receptor grafted T cells is enhanced by anti-idiotypic monoclonal antibodies directed to the scFv domain of the receptor. J Invest Dermatol 1999; 112(5): 744–750.
- Schmidt P, Kopecky C, Hombach A, et al.: Eradication of melanomas by targeted elimination of a minor subset of tumor cells. Proc Natl Acad Sci U S A 2011; 108(6): 2474-2479.
- Harrer D, Simon B, Fujii SI, et al.: RNA-transfection of γ/δ T cells with a chimeric antigen receptor or an α/β T-cell receptor: a safer alternative to genetically engineered α/β T cells for the immunotherapy of melanoma. BMC Cancer 2017; 17: 551.
- Bumol TF, Reisfeld RA.: Unique glycoprotein-proteoglycan complex defined by monoclonal antibody on human melanoma cells. Proc Natl Acad Sci USA 1982; 79(4): 1245–1249.
- Ilieva KM, Cheung A, Mele S, et al.: Chondroitin Sulfate Proteoglycan 4 and Its Potential As an Antibody Immunotherapy Target across Different Tumor Types. Front Immunol 2017; 8: 1911.
- Schiffer D, Mellai M, Boldorini R, et al.: The Significance of Chondroitin Sulfate Proteoglycan 4 (CSPG4) in Human Gliomas. Int J Mol Sci 2018; 19(9).
- Van Sinderen M, Cuman C, Winship A, et al.: The chrondroitin sulfate proteoglycan (CSPG4) regulates human trophoblast function. Placenta 2013; 34(10): 907–912.
- Fukushi J, Makagiansar IT, Stallcup WB: NG2 proteoglycan promotes endothelial cell motility and angiogenesis via engagement of galectin-3 and alpha3beta1 integrin. Mol Biol Cell 2004; 15(8): 3580–3590.
- Sakry D, Neitz A, Singh J, et al.: Oligodendrocyte precursor cells modulate the neuronal network by activity-dependent ectodomain cleavage of glial NG2. PLoS Biol 2014; 12(11): e1001993.
- Legg J, Jensen UB, Broad S, et al.: Role of melanoma chondroitin sulphate proteoglycan in patterning stem cells in human interfollicular epidermis. Development 2003; 130(24): 6049–6063.
- Ferrone S, Chen ZJ, Liu CC, et al.: Human high molecular weight-melanoma associated antigen mimicry by mouse anti-idiotypic monoclonal antibodies MK2-23. Experimental studies and clinical trials in patients with malignant melanoma. Pharmacol Ther 1993; 57(2-3): 259–290.
- Schlingemann RO, Rietveld FJ, de Waal RM, et al.: Expression of the high molecular weight melanoma-associated antigen by pericytes during angiogenesis in tumors and in healing wounds. Am J Pathol 1990; 136(6): 1393–1405.
- Midwood KS, Salter DM: Expression of NG2/human melanoma proteoglycan in human adult articular chondrocytes. Osteoarthritis Cartilage 1998; 6(5): 297–305.
- Tordsson JM, Ohlsson LG, Abrahmsen LB, et al.: Phage-selected primate antibodies fused to superantigens for immunotherapy of malignant melanoma. Cancer Immunol Immunother 2000; 48(12): 691–702.
- Petrini S, Tessa A, Carrozzo R, et al.: Human melanoma/NG2 chondroitin sulfate proteoglycan is expressed in the sarcolemma of postnatal human skeletal myofibers. Abnormal expression in merosin-negative and Duchenne muscular dystrophies. Mol Cell Neurosci 2003; 23(2): 219–231.
- Campoli MR, Chang CC, Kageshita T, et al.: Human high molecular weight-melanoma-associated antigen (HMW-MAA): a melanoma cell surface chondroitin sulfate proteoglycan (MSCP) with biological and clinical significance. Crit Rev Immunol 2004; 24(4): 267–296.
- Beard RE, Abate-Daga D, Rosati SF, et al.: Gene expression profiling using nanostring digital RNA counting to identify potential target antigens for melanoma immunotherapy. Clin Cancer Res 2013; 19(18): 4941–4950.
- Erfurt C, Sun Z, Haendle I, et al.: Tumor-reactive CD4+ T cell responses to the melanoma-associated chondroitin sulphate proteoglycan in melanoma patients and healthy individuals in the absence of autoimmunity. J Immunol 2007; 178(12): 7703–7709.
- Natali PG, Giacomini P, Russo C, et al.: Antigenic profile of human melanoma cells. Analysis with monoclonal antibodies to histocompatibility antigens and to melanoma-associated antigens. J Cutan Pathol 1983; 10(4): 225–237.
- Berd D, Herlyn M, Koprowski H, Mastrangelo MJ: Flow cytometric determination of the frequency and heterogeneity of expression of human melanoma-associated antigens. Cancer Res 1989; 49(23): 6840–6844.
- Morgan AC Jr., Galloway DR, Reisfeld RA: Production and characterization of monoclonal antibody to a melanoma specific glycoprotein. Hybridoma 1981; 1(1): 27–36.
- Morgan AC Jr., Woodhouse C, Bartholemew R, Schroff R: Human melanoma-associated antigens: analysis of antigenic heterogeneity by molecular, serologic and flow-cytometric approaches. Mol Immunol 1986; 23(2): 193–200.
- Li Y, Madigan MC, Lai K, et al.: Human uveal melanoma expresses NG2 immunoreactivity. Br J Ophthalmol 2003; 87(5): 629–632.
- Li Y, Wang J, Rizvi SM, Jager MJ, et al.: In vitro targeting of NG2 antigen by 213Bi-9.2.27 alpha-immunoconjugate induces cytotoxicity in human uveal melanoma cells. Invest Ophthalmol Vis Sci 2005; 46(12): 4365–4371.
- Chekenya M, Rooprai HK, Davies D, et al.: The NG2 chondroitin sulfate proteoglycan: role in malignant progression of human brain tumours. Int J Dev Neurosci 1999; 17(5–6): 421–435.
- Godal A, Bruland O, Haug E, et al.: Unexpected expression of the 250 kD melanoma-associated antigen in human sarcoma cells. Br J Cancer 1986; 53(6): 839–841.
- Shoshan Y, Nishiyama A, Chang A, et al.: Expression of oligodendrocyte progenitor cell antigens by gliomas: implications for the histogenesis of brain tumors. Proc Natl Acad Sci U S A 1999; 96(18): 10361–10366.
- Yadavilli S, Hwang EI, Packer RJ, Nazarian J: The Role of NG2 Proteoglycan in Glioma. Transl Oncol 2016; 9(1): 57–63.
- Behm FG, Smith FO, Raimondi SC, et al.: Human homologue of the rat chondroitin sulfate proteoglycan, NG2, detected by monoclonal antibody 7.1, identifies childhood acute lymphoblastic leukemias with t(4;11)(q21;q23) or t(11;19)(q23;p13) and MLL gene rearrangements. Blood 1996; 87(3): 1134–1139.
- Hilden JM, Smith FO, Frestedt JL, et al.: MLL gene rearrangement, cytogenetic 11q23 abnormalities, and expression of the NG2 molecule in infant acute myeloid leukemia. Blood 1997; 89(10): 3801–3805.
- Schwartz S, Rieder H, Schlager B, et al.: Expression of the human homologue of rat NG2 in adult acute lymphoblastic leukemia: close association with MLL rearrangement and a CD10(-)/CD24(-)/CD65s(+)/CD15(+) B-cell phenotype. Leukemia 2003; 17(8): 1589–1595.
- Smith FO, Rauch C, Williams DE, et al.: The human homologue of rat NG2, a chondroitin sulfate proteoglycan, is not expressed on the cell surface of normal hematopoietic cells but is expressed by acute myeloid leukemia blasts from poor-prognosis patients with abnormalities of chromosome band 11q23. Blood 1996; 87(3): 1123–1133.
- Wuchter C, Harbott J, Schoch C, et al.: Detection of acute leukemia cells with mixed lineage leukemia (MLL) gene rearrangements by flow cytometry using monoclonal antibody 7.1. Leukemia 2000; 14(7): 1232–1238.
- Wang X, Osada T, Wang Y, et al.: CSPG4 protein as a new target for the antibody-based immunotherapy of triple-negative breast cancer. J Natl Cancer Inst 2010; 102(19): 1496–1512.
- Nicolosi PA, Dallatomasina A, Perris R: Theranostic impact of NG2/CSPG4 proteoglycan in cancer. Theranostics 2015; 5(5): 530–544.
- de Vries JE, Keizer GD, te Velde AA, et al.: Characterization of melanoma-associated surface antigens involved in the adhesion and motility of human melanoma cells. Int J Cancer 1986; 38(4): 465–473.
- Ozerdem U: Targeting pericytes diminishes neovascularization in orthotopic uveal melanoma in nerve/glial antigen 2 proteoglycan knockout mouse. Ophthalmic Res 2006; 38(5): 251–254.
- Ozerdem U: Targeting of pericytes diminishes neovascularization and lymphangiogenesis in prostate cancer. Prostate 2006; 66(3): 294–304.
- Ampofo E, Schmitt BM, Menger MD, Laschke MW: The regulatory mechanisms of NG2/CSPG4 expression. Cell Mol Biol Lett 2017; 22: 4.
- Stoiber S, Cadilha BL, Benmebarek MR, et al.: Limitations in the Design of Chimeric Antigen Receptors for Cancer Therapy. Cells 2019; 8(5).
- Sievers NM, Dörrie J, Schaft N: CARs: Beyond T Cells and T Cell-Derived Signaling Domains. Int J Mol Sci 2020; 21(10).
- Harrer DC, Dörrie J, Schaft N: Chimeric Antigen Receptors in Different Cell Types: New Vehicles Join the Race. Hum Gene Ther 2018; 29(5): 547–558.
- Montagner IM, Penna A, Fracasso G, et al.: Anti-PSMA CAR-engineered NK-92 Cells: An Off-the-shelf Cell Therapy for Prostate Cancer. Cells 2020; 9(6).
- Van Cutsem E, Machiels J, Van den Eynde M, et al.: SO-009 – Phase 1 studies assessing the safety and clinical activity of autologous and allogeneic NKG2D-based CAR-T therapy in metastatic colorectal cancer. Annals of Oncology 2019; 30: iv124–iv125.
- Santoro SP, Kim S, Motz GT, et al.: T cells bearing a chimeric antigen receptor against prostate-specific membrane antigen mediate vascular disruption and result in tumor regression. Cancer Immunol Res 2015; 3(1): 68–84.
- Bansal R, Reshef R: Revving the CAR – Combination strategies to enhance CAR T cell effectiveness. Blood Reviews 2020: 100695.
- Harrer DC, Dorrie J, Schaft N: CARs and Drugs: Pharmacological Ways of Boosting CAR-T-Cell Therapy. Int J Mol Sci 2023; 24(3).
DERMATOLOGIE PRAXIS 2024; 19(1): 14–24









