O termo linfomas indolentes cobre um número crescente de linfomas de baixo maligno não-Hodgkin e leucemias predominantemente da série de células B. São diferenciados e classificados uns dos outros com base na sua constelação de marcadores e no seu perfil genético. O tratamento depende da extensão ou da dinâmica da doença e dos sintomas do doente. Existe actualmente uma mudança na terapia de quimioterapia clássica para inibidores de sinal e imunoterapêutica. O principal objectivo no tratamento é muitas vezes controlar a doença durante o máximo de tempo possível com uma tolerância aceitável da terapia.
O termo linfoma indolente não-Hodgkin (iNHL) abrange um grupo biologicamente heterogéneo de linfomas de células maduras e pequenas da série de células B com tendência para a manifestação leucémica. No passado, o termo “indolente” descrevia um grupo de linfomas de crescimento lento e “pouco malignos”, cujos subtipos eram frequentemente difíceis de separar uns dos outros e que nem sempre precisavam de ser diferenciados uns dos outros devido à falta de opções terapêuticas.

Entretanto, os dados imunofenotípicos e moleculares expandidos permitem uma melhor subdivisão em entidades linfoma, que deve ser feita de acordo com a actual classificação de linfoma da OMS 2008 (Tab. 1) [1]. Há iNHL que se transformam em linfoma blástico altamente maligno ou agressivo no decurso da doença, primeiro e principalmente linfoma folicular, seguido de leucemia linfocítica crónica (CLL). Além disso, existem entidades como o linfoma de células do manto (LMC), a maioria das quais (aproximadamente 90% de todos os casos) apresenta um comportamento de crescimento agressivo e são tratadas através de imuno-chemoterapia intensiva (se necessário com terapia de alta dose e substituição autóloga de células estaminais). Mas há também um subgrupo indolente que se manifesta principalmente em pacientes mais velhos com envolvimento de medula óssea e baço. Não esquecer o típico NHL que pode ocorrer num contexto clínico específico como o MALT associado ao Helicobacter pylori-associado ou linfoma de zona marginal associado ao HCV.
Diagnóstico e subtipos
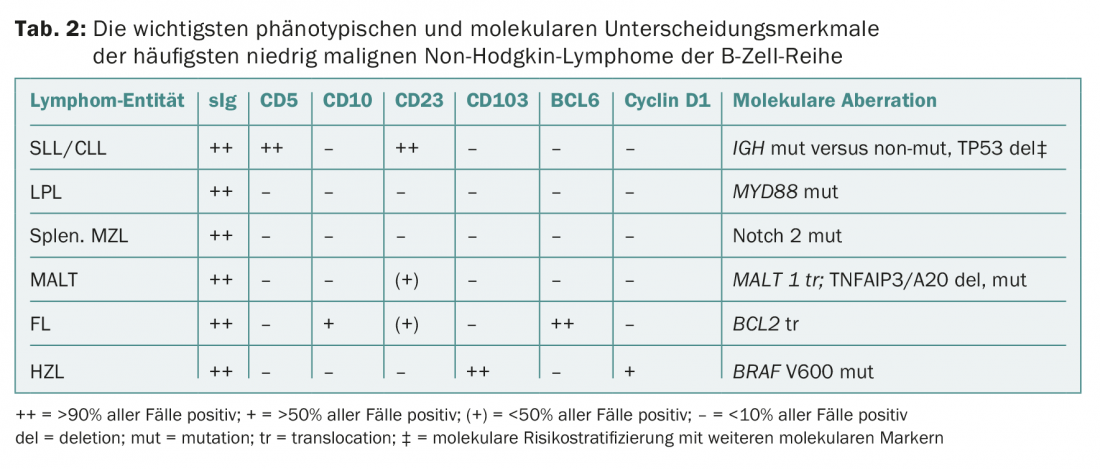
O diagnóstico é feito por biopsia de gânglios linfáticos e/ou medula óssea, fenotipagem (imune) do sangue e/ou medula óssea e análises genético-moleculares (Tab. 2) . Estes testes são normalizados e aplicam-se a todas as patologias do linfoma. De acordo com a classificação da OMS, as seguintes entidades pertencem à iNHL:
- Leucemia linfocítica crónica (CLL) resp. a sua forma nodal (linfoma linfocítico de pequenas células, SLL)
- Linfoma linfo-plasmático (LPL ou doença de Waldenström)
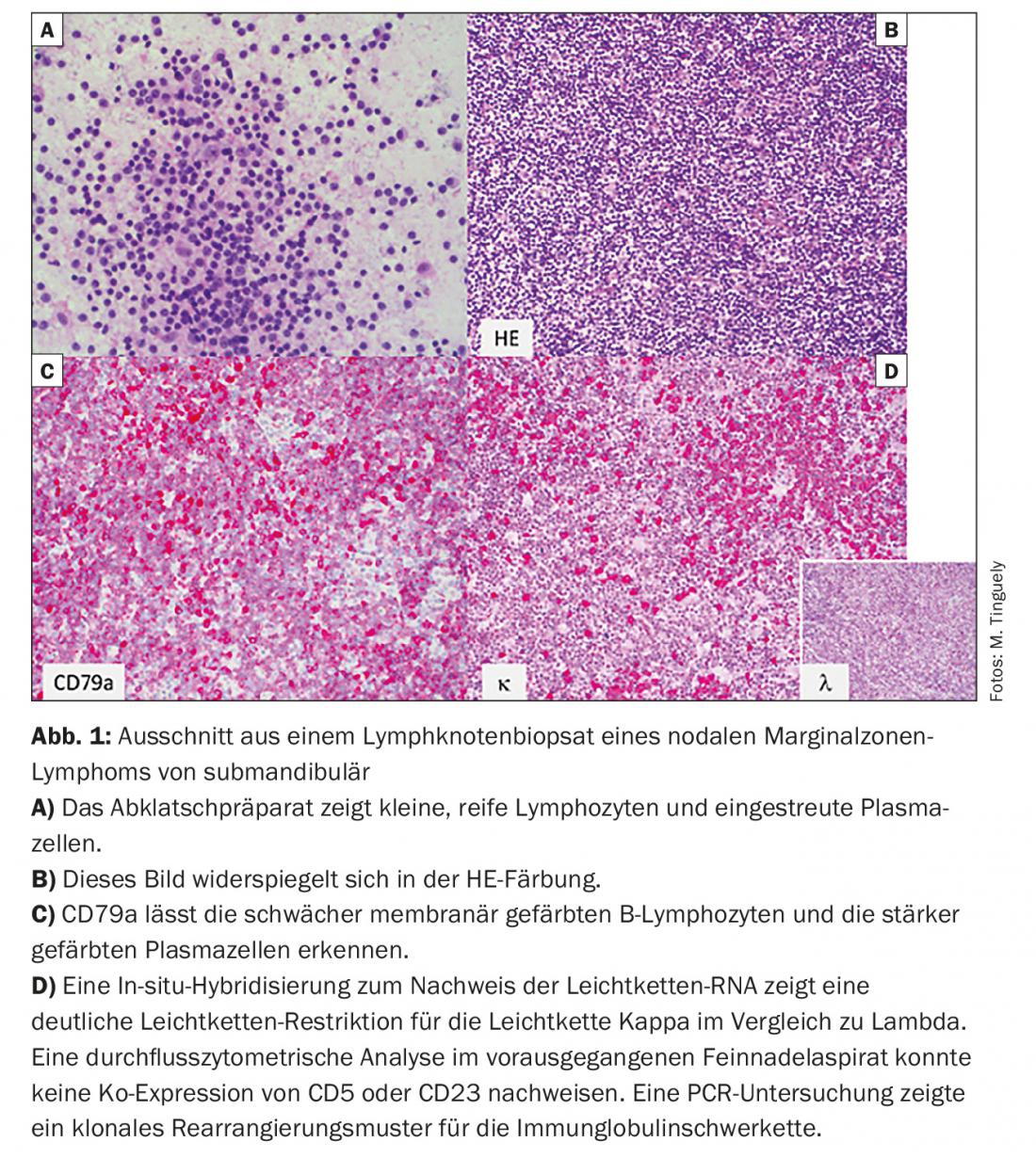
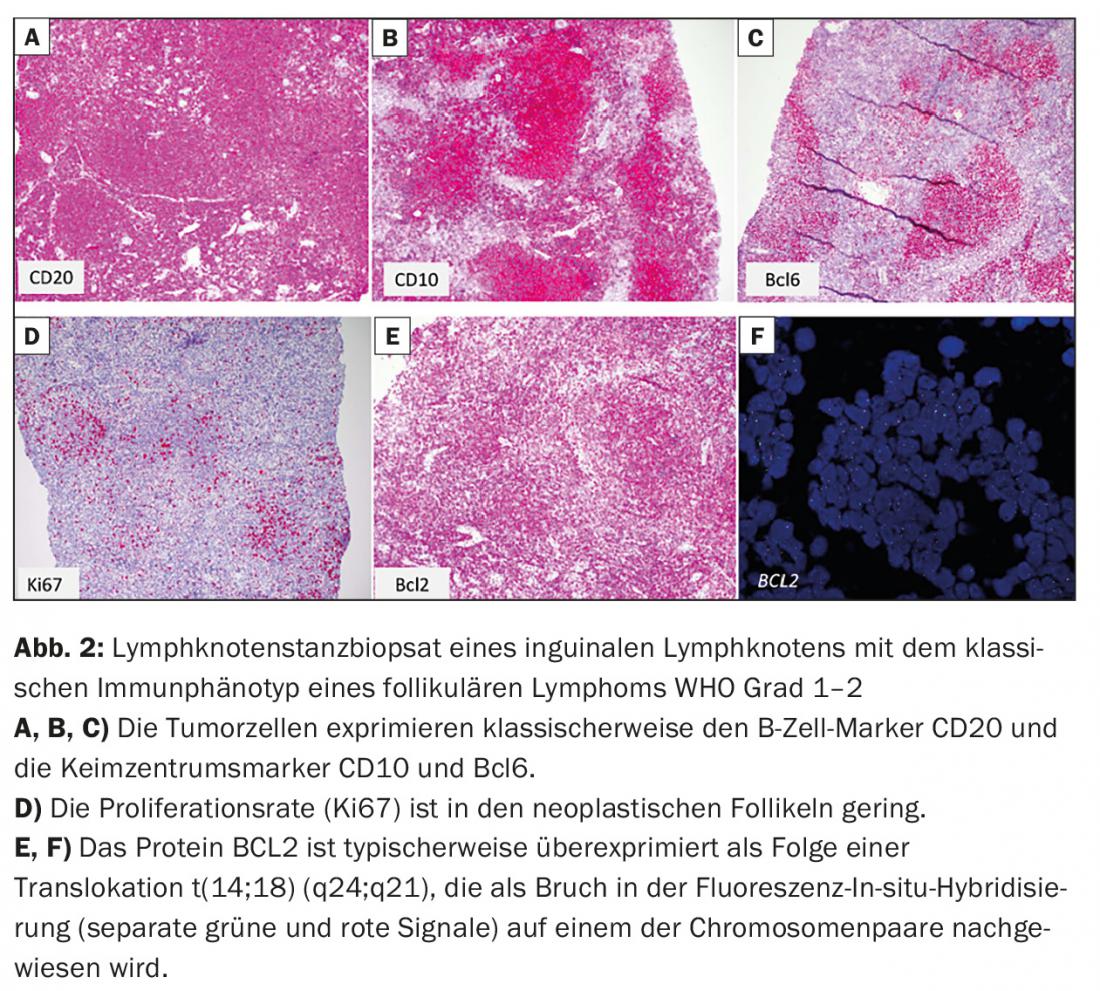
- Linfoma da zona marginal (MZL) (Fig. 1) com os seus três subtipos: nodal, extranodal (por exemplo, como MALT) e esplénico
- Linfoma folicular (FL) (Fig. 2)
- Leucemia de células pilosas (HZL).
A leucemia CLL e a leucemia de células pilosas não são consideradas por todos os grupos de trabalho como fazendo parte da iNHL porque, como o seu nome sugere, muitas vezes manifestam-se principalmente com alterações no hemograma e em alguns casos (isto aplica-se em particular à leucemia de células pilosas) requerem outras medidas terapêuticas. O linfoma de células mantélicas (MCL) já não deve ser contado como iNHL pelas razões mencionadas, embora exista um subgrupo indolente em pacientes mais velhos.
Apresentação clínica e princípios terapêuticos
Os linfomas indolentes são geralmente uma doença de idade avançada. A grande maioria dos pacientes (66%) já se encontra nos estádios avançados III e IV (estadiamento de acordo com Ann Arbor ou, mais recentemente, classificação de Lugano) no momento do diagnóstico e, portanto, já não são passíveis de terapia curativa [2]. O diagnóstico nem sempre está associado à necessidade de iniciar imediatamente a terapia. Embora as recomendações variem um pouco nos subtipos individuais, continua a ser verdade que apenas as doenças sintomáticas devem ser tratadas. A definição de doença sintomática permaneceu em grande parte inalterada nos últimos 30 anos e inclui sintomas locais devido ao crescimento do linfoma, diminuição da função normal dos órgãos (por exemplo, anemia sintomática), sintomas B, doença extranodal sintomática, outras citopénias ou uma taxa de crescimento rápido de uma manifestação de linfoma. Estes critérios são muito elásticos e oferecem ao paciente e ao médico uma ampla margem para a tomada de decisões. Por conseguinte, estão actualmente em curso esforços para estabelecer uma melhor classificação prognóstica e, consequentemente, apoio à decisão através de novos marcadores (moleculares). Um exemplo é o recentemente apresentado índice prognóstico CLL-IPI (CLL-IPI), que fornece uma recomendação sobre o momento e o tipo de terapia [3].
Radioterapia nas fases iniciais: uma opção curativa?
A radioterapia tem sido definida nas últimas décadas como uma opção de tratamento curativo para as fases iniciais do linfoma folicular. 15-25% de todos os doentes diagnosticados com linfoma folicular presente na fase I ou II e as directrizes nacionais e internacionais actuais ainda recomendam a radioterapia para estes doentes [4].
A dificuldade de uma recomendação positiva para a implementação da radioterapia na rotina clínica actual baseia-se na incerteza se os dados de estudos antigos (superfícies de irradiação de grandes áreas, doses totais elevadas, aplicações específicas de campo não linfático ou involuntário) podem ser transferidos para as técnicas de irradiação actuais. Isto coloca o grande dilema de não sermos actualmente capazes de definir o suposto valor curativo da radioterapia para linfomas indolentes (principalmente linfomas foliculares) nas fases iniciais.
Esperar após o diagnóstico: Ainda é válido?
Alguns autores assumem que actualmente cerca de 50% dos pacientes ainda não necessitam de tratamento imediato no momento do diagnóstico. O termo “assistir e esperar” (w&w) foi cunhado há mais de 35 anos [5]. A espera parecia justificável quando a doença progredia lentamente e não havia sintomas ou havia apenas sintomas menores – e também porque não existiam terapias eficazes. Nessa altura, o tempo médio para a primeira terapia para pacientes com FL era de 31-36 meses. As análises observacionais longitudinais mostraram que cerca de 20% dos doentes com FL não necessitavam de terapia com um seguimento mediano de 17 anos. Em comparação com uma coorte de pacientes tratados com quimioterapia no momento do diagnóstico, não houve diferença na sobrevida global (SO) de 5 anos. O SO mediano tinha onze anos e variava muito entre histologias.
Se w&w é apropriado afecta todo o iNHL, mas é analisado principalmente no FL. Assim, num estudo multicêntrico, 379 pacientes foram aleatorizados em três braços de tratamento: apenas observação (w&w), apenas indução rituximab (aplicações de quatro semanas) ou indução rituximab seguida de dois anos de terapia de manutenção (rituximab de dois em dois meses) [6]. Isto mostrou uma vantagem significativa na sobrevivência sem progressão (PFS) para ambos os braços rituximab em comparação com os w&w. No entanto, a sobrevivência global não diferiu, de modo que ainda hoje não existe uma razão convincente para o uso precoce da imunoterapia. Isto pode mudar com novas terapêuticas (imunitárias) e deve, portanto, ser sempre questionado.
Terapias actuais
As opções de tratamento para o iNHL mudaram fundamentalmente nos últimos anos com o estabelecimento de novas terapias. Estes são principalmente baseados em anticorpos e inibição da tirosina quinase (Fig. 3) . Até agora, a imunoterapia com anticorpos específicos CD20 como monoterapia ou em combinação com agentes quimioterápicos clássicos tem dominado a terapia de primeira linha. Deve ainda assumir-se que uma cura não é possível apesar dos métodos terapêuticos modernos; a única excepção é o transplante alogénico de células estaminais.
Anticorpos monoclonais específicos CD20
Um exemplo clássico de um anticorpo específico CD20 é o rituximab, que é agora parte integrante do tratamento do linfoma de células B e pode ser utilizado para várias entidades quer como monoterapia (por exemplo FL) quer em combinação com quimioterapia (por exemplo CLL). Os novos anticorpos anti-CD20 (ex. obinutuzumab) têm uma toxicidade celular directa (actividade ADCC) ainda mais elevada e podem assim eliminar as células linfoma de forma mais eficiente [7]. No tratamento CLL, por exemplo, isto leva a um aumento dos pacientes com doença residual mínima negativa (DRM). Isto significa que em doentes com obinutuzumab + quimioterapia, a doença – em comparação com o tratamento convencional com rituximab + quimioterapia – é mais frequentemente indetectável (38 a 3% no sangue periférico), apesar dos métodos de detecção sensíveis. Resta saber se o aumento da taxa de negatividade do MRD também conduzirá a uma sobrevivência prolongada.
Combinação de anticorpos e quimioterapia
Tipicamente, os anticorpos específicos CD20 são utilizados em combinação com clorambucil, bendamustina ou CHOP. O clorambucil é frequentemente utilizado em combinação com obinutuzumab no tratamento de pacientes CLL mais velhos ou rituximab no linfoma MALT [8]. A bendamustina, por outro lado, em combinação com o rituximab, é considerada o padrão de primeira linha no linfoma folicular (graus 1 e 2) [9] e CLL (com reservas sobre o fludarabine-endoxan). O Rituximab com CHOP é utilizado um pouco menos frequentemente do que no passado e é utilizado principalmente para variantes de linfoma blastóide ou FL grau 3 . Em geral, o número de ciclos de quimioterapia é frequentemente limitado a seis (de três em três ou de quatro em quatro semanas) e a imunoterapia é dada durante o mesmo período de tempo ou como monoterapia durante dois anos como terapia de manutenção.
Imunomoduladores
Os imunomoduladores (IMID) são pequenas moléculas que são geralmente tomadas oralmente. IMIDs como a lenalidomida foram inicialmente utilizadas com sucesso no mieloma múltiplo, e estão agora também a ser testadas no linfoma com taxas de resposta encorajadoras. A combinação de lenalidomida e rituximab em doentes com FL grau 3 e doença recidivante ou refractária atingiu uma taxa de resposta global (ORR) de até 86%. Quando a combinação é utilizada directamente na terapia de primeira linha, os ORRs de até 98% podem ser alcançados com altas taxas de remissão completa (CR) (87% CR e CR não confirmada) e negatividade de MRD. Recentemente, foi também concedida aprovação para o tratamento do linfoma de células mantélicas (MCL). A aprovação é baseada em estudos com doença de MCL recaída ou refractária devido a um ORR de 42-53% [10,11].
Inibição da via de sinalização do receptor da célula B (BCR)
Taxas de resposta ainda mais elevadas no tratamento com MCL podem ser alcançadas por compostos que inibem a via de sinalização BCR a jusante, por exemplo, inibidores de Bruton tirosina cinase (BTK) ou PI3 kinase (PI3K) [12]. Ambas as quinases são frequentemente constitutivamente activas em células linfoma e promovem a proliferação ou sobrevivência celular.
Inibidores BTK
O Ibrutinibe é o primeiro inibidor de BTK aprovado que se liga irreversivelmente covalentemente a um resíduo de cisteína (Cys-481) de tirosina quinase, causando uma forte e sustentada inibição da actividade enzimática. No CLL, o ibrutinibe mostrou uma actividade elevada. A aprovação baseia-se num estudo comparativo com o anticorpo monoclonal específico CD20 do catumumab (estudo RESONATE) em 391 doentes CLL/SLL pré-tratados [13]. Com um seguimento médio de 9,4 meses, o ibrutinibe (420 mg/d/po) melhorou significativamente o PFS e o OS. Após doze meses, o OS era 90% no grupo ibrutinibe e 81% no grupo ofatumumab. A taxa de resposta global foi significativamente mais elevada para o ibrutinibe (42,6 vs. 4,1%, p <0,001). A taxa de resposta e a duração foram independentes da presença de del17p ou da resistência aos análogos purínicos. Isto prova o elevado valor dos inibidores BTK neste subgrupo de pacientes CLL difíceis de tratar. Os acontecimentos adversos mais comuns foram a diarreia, fadiga, febre e náuseas.
A segunda entidade aprovada na Suíça diz respeito ao tratamento de MCL recaídas. A base para a aprovação foi um ensaio multicêntrico, fase II de um braço com 111 pacientes de MCL pré-tratados com uma dose de 560 mg de ibrutinib uma vez por dia. A publicação completa reporta um ORR de 66% com uma taxa de RC de 17% e uma duração média de resposta (DOR) de 17,5 meses [4]. Curiosamente, a taxa de resposta aumentou continuamente no decurso do tratamento (a chamada “resposta incremental sob tratamento”), de modo que – em contraste com as quimioterapias clássicas – também podem ocorrer remissões tardias com terapia contínua.
Inibidores PI3K
A família PI3K é constituída por um número de quinases de serina/treonina que regulam o crescimento, diferenciação, metabolismo, sobrevivência e proliferação em várias células. A inibição da unidade p110δ, por exemplo, leva ao esgotamento significativo das células B e ao bloqueio do caminho de sinalização a jusante do BCR. Por conseguinte, a maioria das abordagens terapêuticas no tratamento do linfoma concentram-se no bloqueio directo da unidade p110δ. O protótipo desta classe de substância é idelalisib como um inibidor selectivo p110δ.
Idelalisib foi inicialmente testado num ensaio aleatório de fase III em 220 doentes com CLL recidivante em combinação com rituximab [15]. No braço de controlo, os pacientes receberam rituximab mais placebo. Com um pré-tratamento mediano com três substâncias, rituximab e um análogo de alquilano ou purina nucleotídica tinham sido utilizados previamente em quase todos os casos. Quase 40% dos doentes CLL também tiveram a alteração genética desfavorável do gene p53.
Em termos de eficácia, houve uma taxa de resposta significativamente mais elevada no braço rituximab + idelalisib (81 vs. 13%), um prolongamento significativo de 1 ano PFS (93 vs. 46%, p<0,001) e um prolongamento significativo de 1 ano OS (92 vs. 80%, p=0,02). A superioridade da combinação de rituximab + idelalisib foi mostrada para todos os subgrupos.
Os efeitos secundários que são significativos para a prática clínica diária e que vão além da aplicação do rituximab são o início precoce ou por vezes tardio da diarreia. No entanto, se se comparar o perfil de efeitos secundários com outras substâncias que poderiam eventualmente ser utilizadas nesta situação, tais como ofatumumab, alemtuzumab ou drogas citostáticas convencionais, o idelalisib certamente pontua positivamente.
Como agente de monoterapia, idelalisib mostra uma elevada eficácia no iNHL no contexto da terapia de recidiva. A substância é aprovada para o tratamento de doentes com FL com doença recidivante que tenham recebido duas linhas anteriores de terapia [16]. No ensaio subjacente da fase II de um braço em 125 doentes iNHL resistentes ao rituximab e aos alquilanos, idelalisib foi administrado a 150 mg duas vezes por dia até à progressão da doença. O tempo médio de resposta foi de 1,9 meses, a duração mediana da resposta foi de 12,5 meses e a mediana do PFS foi de 11 meses. Os eventos adversos de grau 3 ou superior mais comuns foram neutropenia (27% dos pacientes), elevação das aminotransferases (13%), diarreia (13%) e pneumonia (7%).
BCL-2 inibidores
A proteína anti-apoptótica BCL-2 é sobreexpressa nas células do linfoma e contribui para a resistência à quimioterapia. Os inibidores de BCL-2 selectivos, administrados oralmente, tais como o venetoclax, param a proliferação de células linfoma e, assim, levam a remissões tumorais em modelos pré-clínicos. Num ensaio clínico inicial de 106 pacientes com LMC (n=28), linfoma folicular (n=29), linfoma difuso de grandes células B (n=41) e outros subtipos de NHL (n=8), a monoterapia de venetoclax mostrou um perfil de segurança aceitável com a dose máxima tolerada de 1200 mg/d [17]. O ORR foi de 44% para todos os subtipos, 78% para MCL e 38% para FL. O PFS mediano foi de 10-14 meses. Os eventos adversos mais comuns relacionados com o tratamento (EA ≥20%) foram náuseas (48%), diarreia (44%), fadiga (41%), diminuição do apetite (21%) e vómitos (21%). De importância é a ocorrência de síndrome de lise tumoral, que, no entanto, ocorreu em dois pacientes sem consequências clínicas.
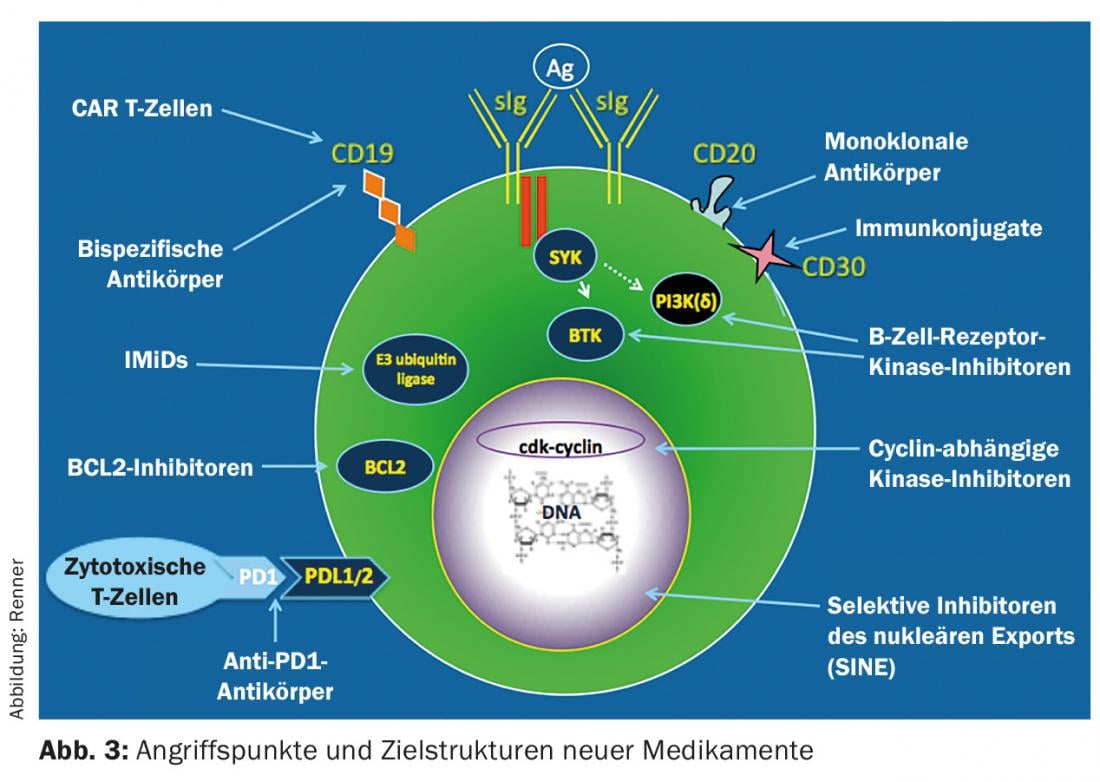
Terapias futuras
As futuras terapias virão principalmente de duas áreas (Fig. 3) : Inibidores da via de sinalização que visam moléculas de comutação importantes da célula linfoma e bloqueiam a sua função (por exemplo, inibidores CDK 4/6), e novas imunoterapêuticas. A constatação de que o sistema imunitário contribui para o controlo de tumores está actualmente a revolucionar as opções de tratamento hemato-oncológico. Os inibidores de bloqueio de pontos de controlo, anticorpos bisespecíficos e células T reprogramadas (células T CAR) estão em desenvolvimento clínico com alguns resultados impressionantes. Talvez um dia consigamos estimular o sistema imunitário de tal forma que seja possível controlar o tumor a longo prazo e até mesmo curá-lo.
Literatura:
- Swerdlow SH, et al.: IARC Press, Lyon, 2008.
- Brice P, et al: J Clin Oncol 1997; 15(3): 1110-1117.
- Molica S, et al: Resumo 498, apresentado na 57ª Reunião Anual da Sociedade Amercan de Hematologia (ASH), 2015.
- Hiddemann W, et al: Internista (Berl) 2016; 57(3): 222-229.
- Morrison VA, Peterson BA: Leuk Lymphoma 1993; 10 Sup: 29-33.
- Adreshna KM, et al: Lancet Oncol 2014; 15(4): 424-435.
- Goede V, et al: NEJM 2014; 370(12): 1101-1110.
- Zucca E, et al: J Clin Oncol 2013; 31(5): 565-572.
- Rummel MJ, et al: Lancet 2013; 381(9873): 1203-1210.
- Habermann TM, et al: Br J Haematol 2009; 145(3): 344-349.
- Witzig TE, et al: Ann Oncol 2011; 22(7): 1622-1627.
- Mato A, et al: Am J Hematol 2015; 90(7): 657-664.
- Byrd JC, et al: NEJM 2014; 371(3): 213-223.
- Wang ML, et al: NEJM 2013; 369(6): 507-516.
- Furman RR, et al: NEJM 2014; 370(11): 997-1007.
- Gopal AK, et al: NEJM 2014; 370(11): 1008-1018.
- Roberts AW, et al: NEJM 2016; 374(4): 311-322.
InFo ONCOLOGy & HaEMATOLOGy 2016; 4(2): 11-15