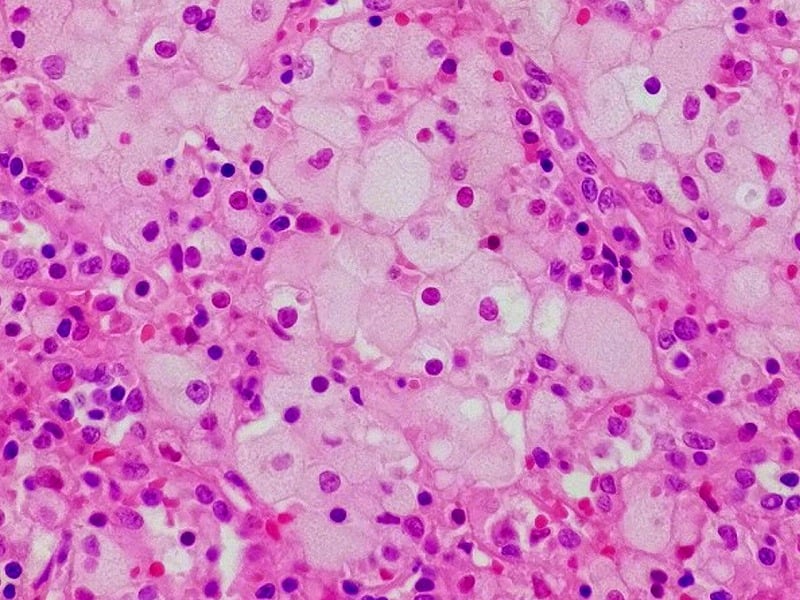
As doenças de depósito lisossomal são um grupo heterogéneo de doenças genéticas que têm origem numa disfunção dos processos metabólicos lisossomais. Atualmente, a doença de Niemann-Pick é também designada por ASMD (“Acid Sphingomeyelinase Deficiency”). Os tipos A e B são classificados como esfingolipidoses, enquanto o tipo C pertence às doenças de armazenamento de lípidos. Para os tipos A e B, a primeira terapia de substituição enzimática foi aprovada na UE no ano passado.
A doença de Niemann-Pick é uma doença genética de armazenamento lisossómico que recebeu o nome do pediatra alemão Albert Niemann (1880-1921) e do patologista alemão Ludwig Pick (1868-1944). Ludwig Pick conseguiu distinguir a doença de Niemann-Pick da doença de Gaucher como uma doença metabólica independente [1]. A substância de armazenamento esfingomielina foi descoberta pelo bioquímico Klenk em 1934. Uma classificação em diferentes subgrupos foi iniciada por Crocker em 1961 [2–4]. As formas mais comuns de manifestação são as do tipo A-C (caixa).
| Os subtipos mais comuns A síndrome de Niemann-Pick é uma doença hereditária autossómica recessiva. Os tipos A e B são causados por uma deficiência na atividade de uma enzima lisossomal codificada pelo gene SMPD1. O defeito genético faz com que a esfingomielina deixe de poder ser decomposta e se acumule nas células de vários órgãos. O tipo C é uma doença do metabolismo do colesterol em que são detectáveis mutações no gene NPC-1 (18q11) ou no gene NPC-2 (14q24.3). |
| para [11] |
Manifestações clínicas
O tipo A é uma doença neurodegenerativa grave da infância que geralmente leva à morte nos primeiros três anos de vida. Os principais sintomas são a hepatoesplenomegalia e o declínio psicomotor. O desenvolvimento das crianças afectadas estagna. As competências aprendidas nos últimos anos de vida perdem-se com o tempo. Frequentemente, podem ser observados problemas de crescimento, vómitos, perda de audição, tetraspasticidade e convulsões mioclónicas antes de completar os primeiros seis meses de vida.
O tipo B caracteriza-se por um início mais tardio da doença e uma manifestação mais ligeira do que o tipo A [5]. A maioria dos doentes atinge a idade adulta e não são observados sintomas cerebrais. Em contrapartida, é caraterística a hepatoesplenomegalia com hiperesplenismo progressivo e disfunção hepática estável, bem como uma deterioração gradual da função pulmonar, acompanhada de osteopenia e de um perfil lipídico aterogénico [6]. Um perfil lipídico pró-aterogénico é observado no início do curso da doença e alguns doentes desenvolvem doença arterial coronária.
O tipo C está associado a uma deficiência no transporte de colesterol dos lisossomas, resultando num aumento do armazenamento de colesterol, gicosfingolípidos e gangliosídeos nos lisossomas de várias células do corpo [7,8]. Em comparação com o tipo A/B, a apresentação clínica é muito heterogénea. Trata-se de uma doença neurovisceral crónica que é mais lentamente progressiva em comparação com o tipo A. Pode ser dividida numa forma infantil precoce, numa forma infantil tardia, numa forma juvenil e numa forma adulta [7]. Clinicamente, as pessoas afectadas apresentam várias anomalias neurológicas e psiquiátricas, por vezes também sintomas viscerais como a hepatoesplenomegalia [8]. As manifestações neurológicas típicas do tipo C são perturbações cognitivas, crises epilépticas, anomalias comportamentais, depressão e psicoses, paralisia do olhar vertical, perturbações da fala e da deglutição e distonia [9].
| Tipo A/B: nova terapêutica de substituição enzimática: olipudase alfa Para doentes pediátricos e adultos com doença de Niemann-Pick ou ASMD (“deficiência de esfingomielinase ácida”) tipo A/B ou tipo B sem envolvimento do sistema nervoso central, a terapia de substituição enzimática olipudase alfa foi aprovada pela EMA em 2022. A olipudase alfa foi concebida para substituir o ASM em falta ou defeituoso para permitir a degradação da esfingomielina. A decisão de aprovação baseia-se nos dados dos ensaios clínicos ASCEND e ASCEND-Peds, que demonstraram melhorias clinicamente relevantes na função pulmonar e uma redução no volume do baço e do fígado quando tratados com olipudase alfa. A incidência de acontecimentos adversos nos doentes que receberam olipudase alfa foi comparável à do grupo do placebo. A Olipudase alfa é administrada por perfusão de quinze em quinze dias na fase de manutenção. No estudo ASCEND, 36 doentes adultos com ASMD tipo A/B ou tipo B foram aleatorizados para receber olipudase alfa ou placebo. Após 52 semanas, o braço de tratamento mostrou uma melhoria na função pulmonar e uma redução do volume do baço. No estudo de braço único ASCEND-PEDS, 20 doentes pediátricos com ASMD tipo A/B ou tipo B foram tratados com olipudase alfa durante 64 semanas. Também neste caso, os parâmetros mais importantes foram atingidos na semana 52. |
| para [12] |
Diagnóstico e terapia
Em culturas de leucócitos e fibroblastos, pode ser detectada a atividade reduzida ou ausente da esfingomielinase ácida, e a causa da doença de Niemann-Pick tipo A e B é [10]. Estes exames genéticos enzimáticos e moleculares podem ser efectuados já no período pré-natal se for conhecida uma predisposição familiar [1]. Para estabelecer o diagnóstico da doença de Niemann-Pick tipo C , devem ser efectuados exames complexos do metabolismo do colesterol [10]. Os defeitos genéticos subjacentes não são atualmente tratáveis (a partir de 2022). A terapêutica de substituição enzimática com olipudase alfa está disponível na UE para os tipos A e B da doença de Niemann-Pick (caixa). O tipo C é tratado sintomaticamente com miglustato.
Literatura:
- “Examination of the liver and spleen by elastography in Niemann-Pick type B disease patients”, Gözde Aksu, dissertação inaugural, 2020, https://openscience.ub.uni-mainz.de,(último acesso em 12.10.2023).
- Crocker AC, Mays VB: Síntese de esfingomielina na doença de Niemann-Pick. Am J Clin Nutr 1961; 9: 63-67.
- Crocker AC: O defeito cerebral na doença de Tay-Sachs e na doença de Niemann-Pick. J Neurochem 1961; 7: 69-80.
- E. K. Sobre a natureza dos fosfatídeos do baço na doença de Niemann-Picksen. Hoppe Seyler’s Journal of Physiological Chemistry. 1934.
- Tran C, et al: Envolvimento pulmonar em pacientes adultos com erros inatos do metabolismo. Karger Compass Pneumol 2018; 6: 6-17.
- Orphanet, www.orpha.net,(último acesso em 12.10.2023)
- Di Lazzaro V, et al: Niemann-Pick tipo C: foco na forma de início na adolescência/adulto. Int J Neurosci 2016; 126(11): 963-971.
- Hammerschmidt TG, et al: Biomarcadores moleculares e bioquímicos para diagnóstico e monitorização da terapia de doentes com Niemann-Pick tipo C. Int J Dev Neurosci 2017; 66: 18-23.
- Bonnot O, et al.: Sintomas psiquiátricos e neurológicos em doentes com doença de Niemann-Pick tipo C (NP-C): Resultados do Registo Internacional de NPC. World J Biol Psychiatry 2017: 1-10.
- “Doença de Niemann-Pick”, https://flexikon.doccheck.com,(último acesso em 12.10.2023)
- Desnick JP, et al.: Identificação e caraterização de oito novas mutações SMPD1 que causam a doença de Niemann Pick tipos A e B. Mol Med 2010; 16: 316-321.
- “Xenpozyme® (olipudase alfa) aprovado pela Comissão Europeia como primeiro e único tratamento para ASMD”, 28.06.2022.
| Imagem da capa: Célula de Niemann no baço. ©W.CC, Wikimedia |
GP PRACTICE 2023; 18(10): 48