O tratamento de escolha para o sarcoma de Ewing inclui quimioterapia neoadjuvante, ressecção tumoral e quimioterapia pós-operatória com/sem radioterapia. Neste contexto, a gestão interdisciplinar num centro apropriado é de importância crucial.
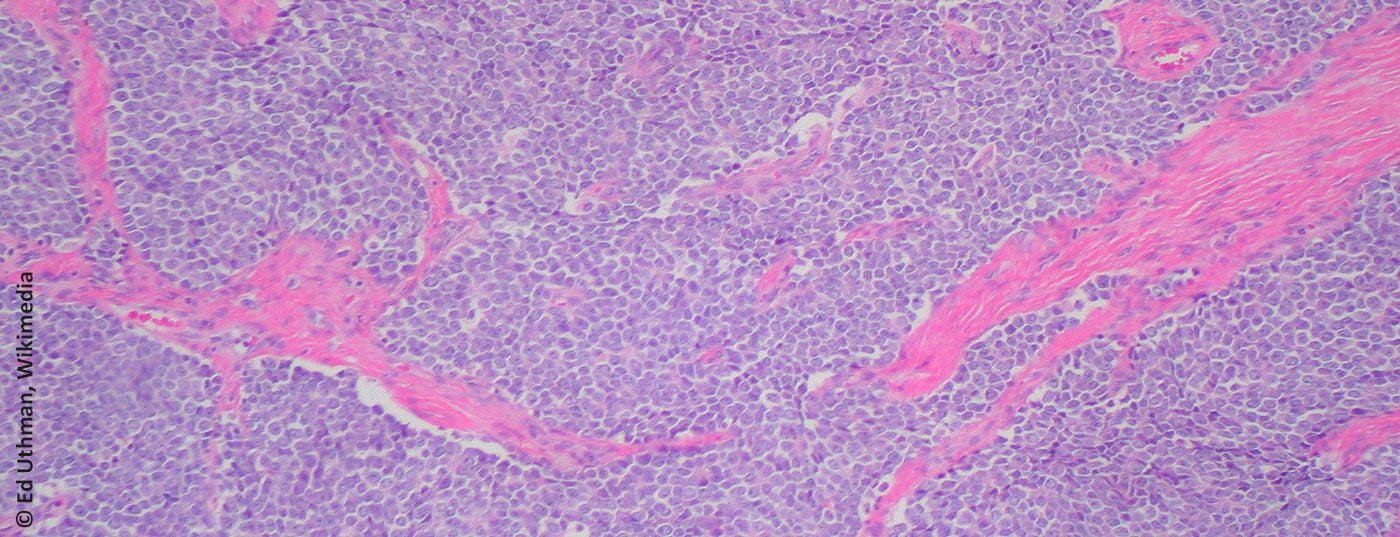
O sarcoma de Ewing recebeu o nome de James Ewing (patologista americano, 1866-1943). Microscopicamente, pertence ao grupo dos pequenos tumores de células azuis e redondas (Fig. 1) . A célula de origem não é claramente compreendida, embora a presença de marcadores neuronais sugira uma ligação com o neuroectodermato embrionário [1]. O diagnóstico é geralmente feito através da detecção de diagnóstico molecular de translocações envolvendo o gene EWS no cromossoma 22. A translocação mais comum (85-95%) é t(11;22)(q24;q12) [2]. Por definição, todos os sarcomas de Ewing são classificados como altamente malignos (G3) [3].
Epidemiologia
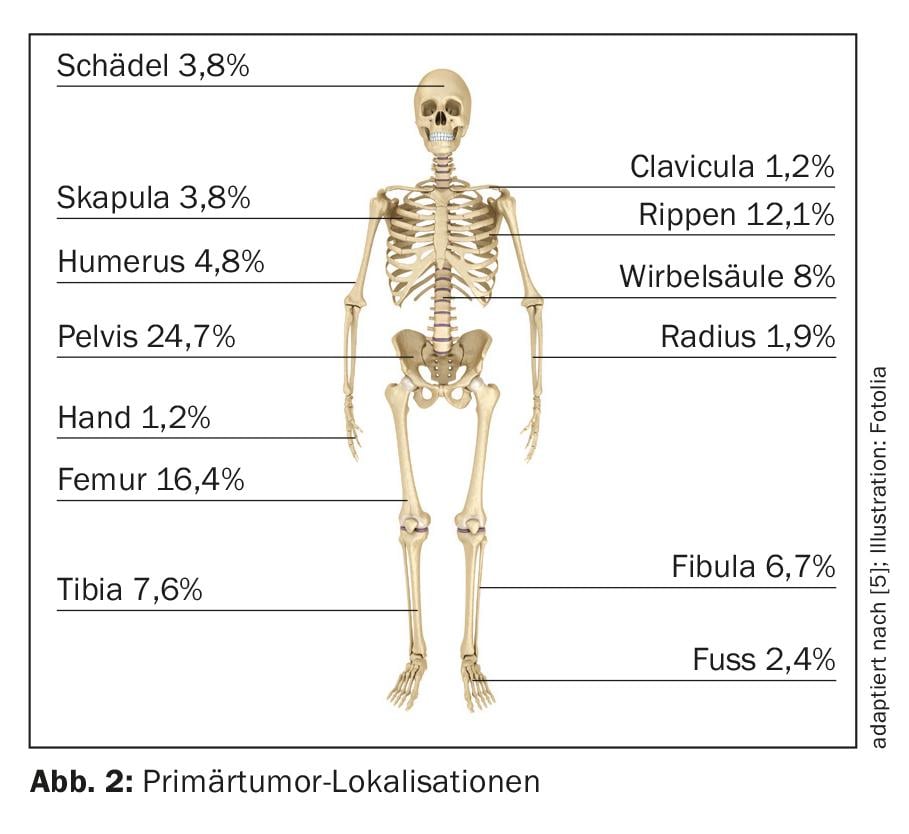
Após o osteosarcoma, o sarcoma de Ewing representa o segundo tumor ósseo maligno primário mais comum na infância e adolescência. A idade média de início é de 10-15 anos, a taxa de incidência anual é de cerca de 3/1’000’000 de população [4]. O sexo masculino é afectado ligeiramente mais frequentemente (1,5:1). A pélvis (25%) e as diafises dos ossos tubulares longos, especialmente no fémur (aproximadamente 16% ) (Fig. 2), estão entre as localizações de tumores primários mais frequentes [5]. Em 15% dos casos, uma localização extra-óssea pode ser principalmente documentada [6].
Clínica
Os sintomas iniciais não são específicos. Uma dor localizada e/ou um inchaço, possivelmente com restrições de mobilidade consecutivas, estão em primeiro plano dos sintomas. Em cerca de 10-15% dos casos, uma fractura patológica está presente no diagnóstico inicial [7], em 80% uma fase de tumor formalmente localizada. Devido a uma taxa de metástases muito elevada (>80%) após terapia exclusivamente local do tumor primário, pode-se assumir em quase todos os casos que as metástases subclínicas já estão presentes [8]. A metástase ocorre mais frequentemente pulmonar, óssea e na medula óssea [9].
Diagnósticos
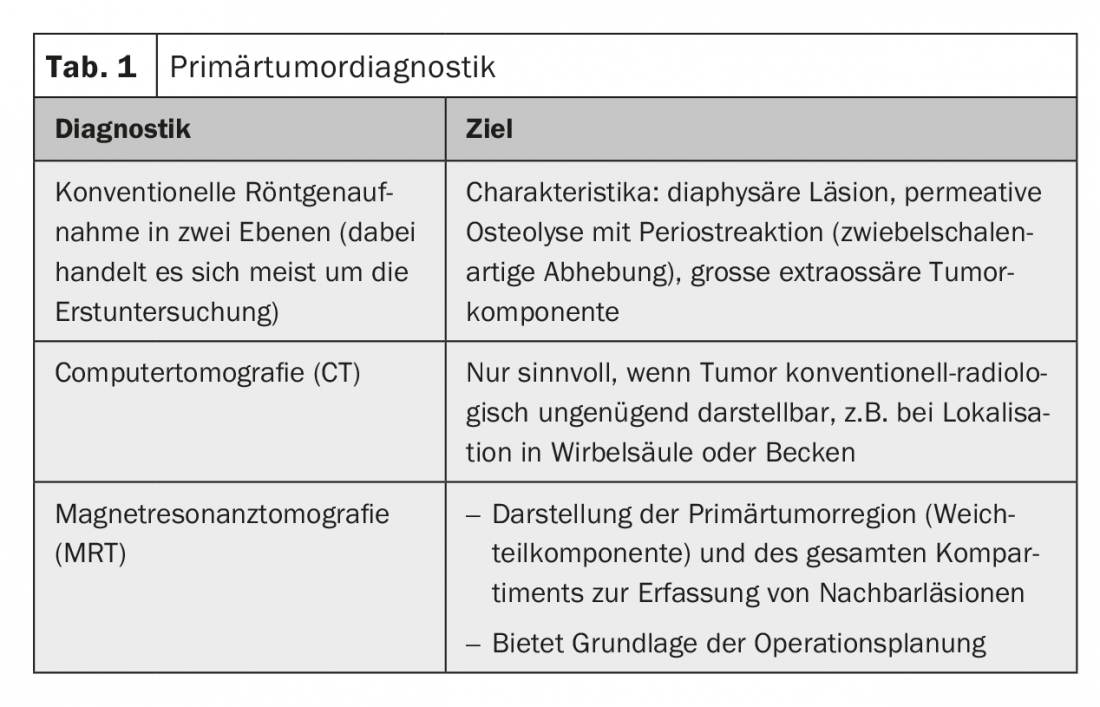
Diagnóstico radiológico do tumor primário: O diagnóstico radiológico do tumor primário deve ser sempre realizado antes da biópsia. É a base para uma avaliação do significado e da resectabilidade do tumor, assim como para o planeamento da biópsia (tab. 1).
Biopsia: Se houver suspeita de um tecido mole maligno ou tumor ósseo, a biopsia é obrigatória para confirmar o diagnóstico. A biopsia deve ser realizada com o envolvimento de um cirurgião experiente no tratamento de sarcomas, idealmente o futuro cirurgião. A via de acesso cirúrgico deve ser aqui tida em conta. O padrão de ouro é a biópsia do núcleo da agulha.
Encenação: Após confirmação da biópsia do diagnóstico, devem ser organizados os seguintes exames de encenação:
- Tórax CT (exclusão das metástases pulmonares)
- Cintilografia esquelética (exclusão das metástases ósseas)
- Biopsia e aspiração de medula óssea (apenas indicada se o PET/CT não for realizado devido à baixa incidência).
- Outros procedimentos de imagiologia dependendo de queixas clínicas.
O valor de um exame PET/CT na fase inicial e como seguimento de imagem está actualmente a ser investigado em ensaios clínicos. A sensibilidade da detecção de metástases pulmonares é menor em comparação com o tórax da TC, a sensibilidade da detecção de lesões ósseas é maior [10,11].
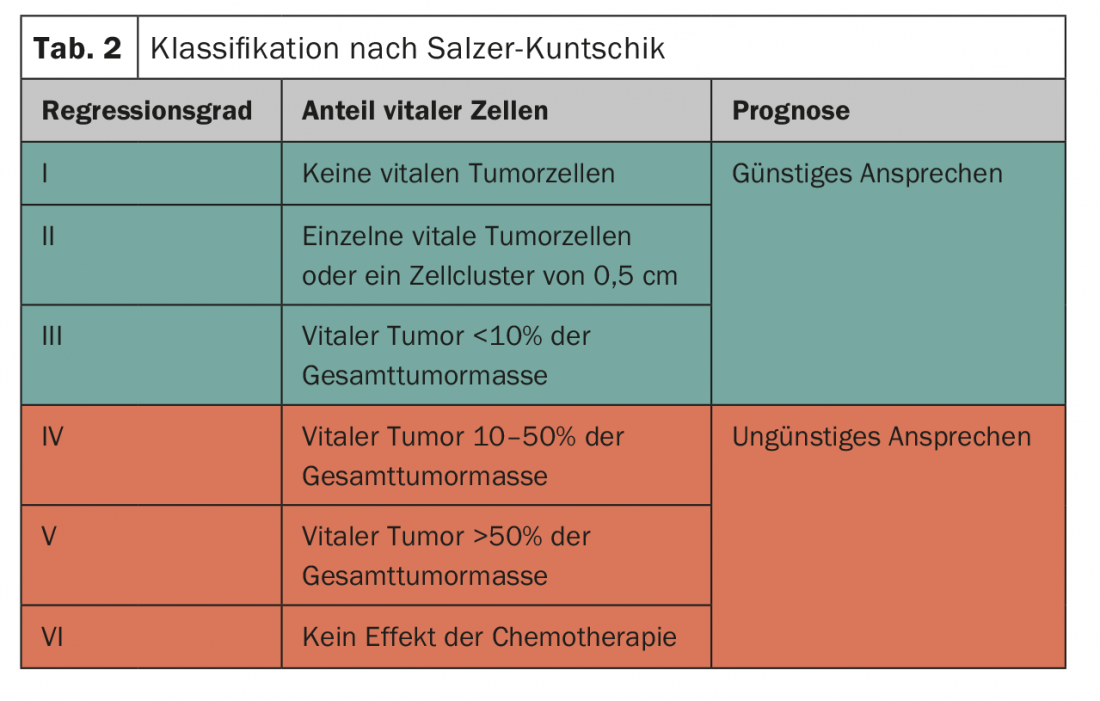
Resposta tumoral histológica à terapia do sistema: Para avaliar a resposta à quimioterapia, a proporção de células malignas vitais é determinada na amostra de ressecção definitiva. O patologista classifica então a resposta nos países de língua alemã com base nesta avaliação (Tab. 2) [12]. A classificação tem um significado prognóstico; é de grande relevância especialmente no contexto dos estudos da Euro-Ewing para determinar a estratégia terapêutica pós-operatória.
Fase localizada da doença
A terapia do sarcoma de Ewing é multimodal e deve ser discutida de uma forma interdisciplinar num centro de sarcoma. Isto inclui sempre terapia local (ressecção e/ou radioterapia) e quimioterapia. Os primeiros ensaios prospectivos aleatórios demonstraram uma sobrevivência global significativamente melhor com quimioterapia adicional (10-20% contra pouco menos de 70% em 5 anos de sobrevivência sem eventos) [8,13], razão pela qual o actual padrão de cuidados inclui quimioterapia neoadjuvante seguida de terapia local do tumor primário e quimioterapia adjuvante [14]. Os pacientes devem ser tratados dentro de um protocolo de estudo, se possível.
Cirurgia: A ressecção do tumor primário é a terapia local de escolha. O objectivo é sempre o de conseguir uma ressecção tumoral completa. No entanto, a ressecabilidade do tumor primário não é por vezes dada em função da localização anatómica. Estes incluem principalmente os sarcomas de Ewing da coluna vertebral e da pélvis. A modalidade terapêutica deve ser individualizada nestes casos. Neste caso, a ressecção com radioterapia pós-operatória ou radioterapia apenas é dirigida a [15,16].
Radioterapia: Como tumores sensíveis à radiação, os sarcomas de Ewing mostram uma taxa de controlo local comparável à da radioterapia como pacientes tratados cirurgicamente em certos estudos [17]. A radioterapia definitiva é preferida para tumores primários não ressecáveis de acordo com uma ressecção incompleta seguida de radioterapia pós-operatória [17]. No caso de excisão marginal/intralesional, há uma indicação para realizar radioterapia pós-operatória. O valor da radioterapia aditiva em casos de resposta histológica insuficiente à quimioterapia (mas ressecção tumoral completa) não é claro. Em princípio, a radioterapia também pode ser administrada pré-operatoriamente [18].
Terapia do sistema: O padrão internacional é a quimioterapia combinada. Os agentes quimioterápicos mais eficazes incluem substâncias alquilantes (ifosfamida, ciclofosfamida), antraciclinas (doxorubicina), bem como etoposida, vincristina e actinomicina. O regime VIDE (vincristina, ifosfamida, doxorubicina, etoposida) em analogia com o protocolo Euro-Ewing 1999 e 2008 na Europa e o regime VDC/IE (agentes VIDE + ciclofosfamida) na América servem frequentemente como modelos para a quimioterapia de indução. A adição de ifosfamida e etoposida à VDC foi associada a um prolongamento significativo de 5 anos de sobrevivência sem eventos no ensaio aleatório IESS-III (69% vs. 54%) [19]. A comparação directa entre os esquemas é testada no estudo mais recente da Euro-Ewing de 2012.
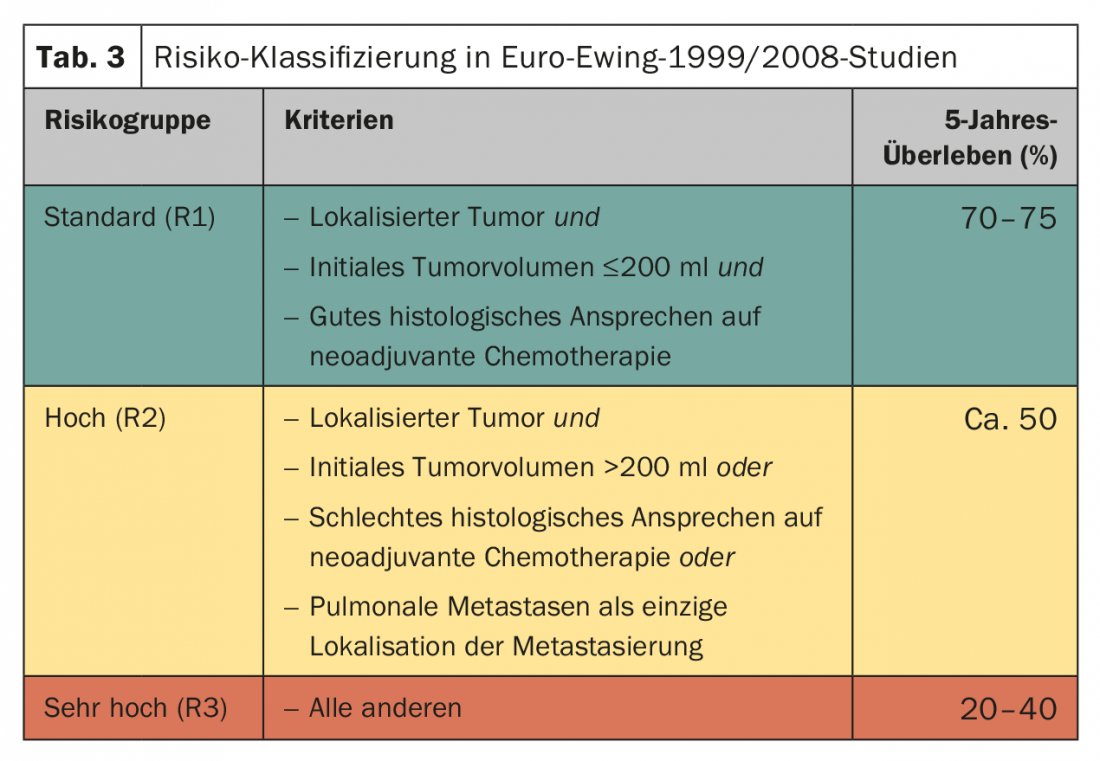
A terapia local segue geralmente seis ciclos de quimioterapia neoadjuvante. A terapia pós-operatória é planeada em função da constelação de risco (tab. 3) . O padrão são oito ciclos de quimioterapia de acordo com VAI ou VAC. O ensaio Euro-Ewing 1999 comparou aleatoriamente a eficácia da VAC (ciclofosfamida) e VAI (ifosfamida) no grupo de risco R1. Basicamente, a equivalência dos regimes foi confirmada, parecendo o sexo masculino beneficiar mais do VAI (HR 1,34; 95% CI 0,96-1,86) [20]. A duração total da terapia é de cerca de dez a doze meses. No grupo de risco R2 (fase localizada do tumor, má resposta histológica do tumor, volume do tumor >200 ml), o significado da terapia de alta dose com melfalanina com transplante de células estaminais autólogas foi aleatorizado. Aqui, uma melhoria significativa na sobrevivência global foi alcançada através de quimioterapia de alta dose com busulfan/melphalan (77,8 vs. 69,9%, HR 0,60 [0,39–0,92], p=0,019) [21].
Fase da doença metástática
Na fase de metástase, o sarcoma de Ewing responde basicamente aos mesmos agentes quimioterápicos que são utilizados na fase de localização. Dependendo do número e especialmente da localização, existe um potencial curativo, apesar da presença de metástases hematogénicas. Para metástases pulmonares/pleurais isoladas, a taxa de cura é de até 40%, para metástases ósseas e da medula óssea é de cerca de 20-25% e para uma combinação destes locais é de 15% [22]. Tais resultados são alcançados em particular com uma terapia local consistente de todas as metástases.
Metástases pulmonares/pleurais: Se as metástases pulmonares persistentes forem ressecadas após a quimioterapia de indução, isto parece estar associado a um prognóstico melhorado [23] – correspondendo à recomendação de remover cirurgicamente metástases pulmonares radiologicamente visíveis durante o curso. Uma vez que são frequentemente vistos mais focos intra-operatórios do que objectivos por estadiamento pré-operatório, a ressecção deve ser realizada por cirurgia aberta [24].
De acordo com análises retrospectivas, a irradiação parenquimatosa pulmonar (entre 15 e 20 Gy) está também associada a um prognóstico mais favorável em casos de metástase pneumo-pleural isolada [25,26]. A indicação deve assim ser avaliada em remissão completa após a quimioterapia, bem como após a ressecção de todos os focos pulmonares.
Metástases ósseas e de medula óssea: No caso de metástases ósseas e/ou da medula óssea, o prognóstico é pobre apesar do objectivo terapêutico potencialmente curativo. No caso de envolvimento oligometástatico, recomenda-se que se considerem opções terapêuticas locais. A radioterapia é o principal método aqui utilizado.
Quimioterapia de alta dose com transplante de células estaminais autólogas: O valor da quimioterapia de alta dose com suporte de células estaminais autólogas tem sido discutido de forma controversa na literatura há muito tempo. Um estudo prospectivo não randomizado mostrou resultados excepcionais com uma sobrevivência livre de eventos de 5 anos até 52%. No entanto, outras publicações não puderam apoiar estes resultados [27,28]. Só recentemente, os resultados da estratificação R2 foram comunicados no contexto do Euro-Ewing 1999. Em doentes com metástases pulmonares, a quimioterapia de alta dose sem irradiação pulmonar não demonstrou um benefício em comparação com a quimioterapia convencional com irradiação pulmonar [29]. Os resultados do grupo de risco R3 ainda estão pendentes.
Tratamento de recidivas
As recidivas mais frequentes são encontradas nos primeiros cinco anos após o diagnóstico inicial, mas as recidivas tardias também não são incomuns [30]. O prognóstico das recidivas nos primeiros dois anos é muito pobre, enquanto as recidivas posteriores mostram uma sobrevivência a longo prazo em cerca de 15-20% [31]. A terapia de escolha depende do tempo da recorrência, da localização e do número de manifestações tumorais e da terapia anterior. As recidivas locais e metástases pulmonares isoladas são geralmente tratadas localmente, ou seja, com ressecção e/ou radioterapia [32]. No caso de focos extensivos de recorrência, é novamente indicado o início da terapia sistémica, embora não exista um regime padrão para tal. Se houver uma boa e, sobretudo, longa resposta à terapia inicial, pode ser avaliada uma repetição da mesma. A dose cumulativa de doxorubicina não deve ser desconsiderada. Existe uma tendência geral para intensificar a quimioterapia através de uma terapia de alta dose, embora as provas a este respeito sejam limitadas [33]. Os regimes de quimioterapia utilizados incluem topotecan/cicloposfamida, irinotecan/temozolomida, gemcitabina/docetaxel, infosfamida de alta dose e quimioterapia com etoposida contendo platina [34–36]. As terapias com objectivos moleculares (por exemplo, inibidores IGF-1 e PARP) bem como as abordagens imunoterapêuticas estão actualmente a ser investigadas em ensaios.
Pós-tratamento
O acompanhamento visa a detecção precoce de recidivas e o controlo de toxicidade tardia. Actualmente, há uma falta de dados prospectivos mostrando um benefício de sobrevivência através de exames regulares de acompanhamento. Os intervalos de seguimento são uma tentativa de fazer justiça ao aumento da probabilidade de recorrência nos primeiros dois a três anos. As recomendações podem ser encontradas, por exemplo, nas directrizes da National Comprehensive Cancer Network (NCCN).
As mais comuns são as neoplasias secundárias (incidência acumulada 9%), endocrinopatias incl. Infertilidade, cardio-, nefro- e neurotoxicidade, toxicidade pulmonar, bem como o comprometimento funcional locoregional no contexto da terapia local como toxicidade terapêutica tardia em [37]. Detalhes e recomendações sobre a monitorização de efeitos tardios podem ser encontrados em www.survivorshipguidelines.org.
Mensagens Take-Home
- O diagnóstico e terapia do sarcoma de Ewing são realizados de forma interdisciplinar num centro de sarcoma. O diagnóstico radiológico do tumor primário deve ser realizado antes da biópsia. O tratamento de escolha é um neoadjuvante
- Quimioterapia, uma ressecção tumoral e quimioterapia pós-operatória com/sem radioterapia.
- O tumor deve ser ressecado por um cirurgião experiente em sarcomoncologia.
- Mesmo na fase de metástase do tumor, o objectivo terapêutico é curativo.
Literatura:
- Lipinski M, et al: Antigénios associados ao neuroectodermas nas linhas celulares do sarcoma de Ewing. Investigação do cancro 1987; 47: 183-187.
- de Alava E, Gerald WL: Biologia molecular do sarcoma de Ewing/ família de tumores neuroectodérmicos primitivos. Journal of clinical oncology: revista oficial da Sociedade Americana de Oncologia Clínica 2000; 18: 204-213.
- Fletcher CDM BJ, Hogendoorn P, Mertens F (eds.): WHO Classification of Tumours of Soft Tissue and Bone (IARC WHO Classification of Tumours). 4ª ed. 2013.
- Esiashvili N, Goodman M, Marcus RB Jr: Alterações na incidência e sobrevivência de doentes com sarcoma de Ewing nas últimas 3 décadas: Epidemiologia de vigilância e dados de resultados finais. Journal of pediatric hematology/oncology 2008; 30: 425-430.
- Cotterill SJ, et al: Factores prognósticos no tumor ósseo de Ewing: análise de 975 pacientes do Grupo de Estudo do Sarcoma da Cooperativa Europeia Intergrupal Ewing. Journal of clinical oncology: revista oficial da Sociedade Americana de Oncologia Clínica 2000; 18: 3108-3114.
- Applebaum MA, et al: Características clínicas e resultados em pacientes com sarcoma de Ewing extraesquelético. Cancro 2011; 117: 3027-3032.
- Widhe B, Widhe T: sintomas iniciais e características clínicas do osteossarcoma e do sarcoma de Ewing. The Journal of bone and joint surgery volume americano 2000; 82: 667-674.
- Nesbit ME Jr, et al: Multimodal therapy for the management of primary, nonmetastatic Ewing’s s sarcoma of bone: a long-term follow-up of the First Intergroup study. Journal of clinical oncology: revista oficial da Sociedade Americana de Oncologia Clínica 1990; 8: 1664-1674.
- Applebaum MA, et al: Características clínicas e resultados em doentes com sarcoma de Ewing e envolvimento de gânglios linfáticos regionais. Sangue pediátrico e cancro 2012; 59: 617-620.
- Franzius C, et al: FDG-PET para a detecção de metástases pulmonares de tumores ósseos primários malignos: comparação com a TC helicoidal. Anais de oncologia: revista oficial da Sociedade Europeia de Oncologia Médica/ESMO 2001; 12: 479-486.
- Franzius C, et al: FDG-PET para a detecção de metástases ósseas de tumores ósseos primários malignos: comparação com cintigrafia óssea. Revista europeia de medicina nuclear 2000; 27: 1305-1311.
- Salzer-Kuntschik M, et al: graus morfológicos de regressão no osteossarcoma após a poli-quimioterapia – estudo COSS 80. Journal of cancer research and clinical oncology 1983; 106 Suppl: 21-24.
- Burgert EO Jr, et al: Multimodal therapy for the management of nonpelvic, localized Ewing’sarcoma of bone: estudo intergrupal IESS-II. Journal of clinical oncology: revista oficial da Sociedade Americana de Oncologia Clínica 1990; 8: 1514-1524.
- Womer RB, et al: Randomized controlled trial of interval-compressed chemotherapy for the treatment of localized Ewing sarcoma: a report from the Children’s Oncology Group. Journal of clinical oncology: revista oficial da Sociedade Americana de Oncologia Clínica 2012; 30: 4148-4154.
- Vogin G, et al: Controlo local e sequelas em tumores de Ewing localizados da coluna vertebral: um estudo retrospectivo francês. Eur J Cancer 2013; 49: 1314-1323.
- Puri A, et al: Resultados da ressecção cirúrgica no sarcoma de Ewing pélvico. Journal of surgical oncology 2012; 106: 417-422.
- La TH, et al: Radiation therapy for Ewing’s s sarcoma: resultados do Memorial Sloan-Kettering na era moderna. International journal of radiation oncology, biology, physics 2006; 64: 544-550.
- Krasin MJ, et al: Irradiação definitiva na gestão multidisciplinar do sarcoma de Ewing localizado família de tumores em pacientes pediátricos: resultado e factores prognósticos. International journal of radiation oncology, biology, physics 2004; 60: 830-838.
- Grier HE, et al: Adição de ifosfamida e etoposida à quimioterapia padrão para sarcoma de Ewing e tumor neuroectodermal primitivo de osso. The New England journal of medicine 2003; 348: 694-701.
- Le Deley MC, et al: Cyclophosphamide comparado com ifosfamide no tratamento de consolidação do sarcoma de Ewing de risco padrão: resultados do ensaio aleatório de não-inferioridade Euro-EWING99-R1. Journal of clinical oncology: revista oficial da Sociedade Americana de Oncologia Clínica 2014; 32: 2440-2448.
- Whelan J, et al: Eficácia da consolidação da quimioterapia de alta dose de busulfan-melphalan (BuMel) no sarcoma de Ewing de alto risco localizado (ES): Resultados do ensaio aleatório EURO-EWING 99-R2 (EE99R2Loc). Journal of Clinical Oncology 2016; 34(15) Suplemento: 11000-11000.
- Paulussen M, et al: Tumores de Ewing com metástases pulmonares primárias: análise de sobrevivência de 114 pacientes da Cooperativa Ewing (Intergrupo Europeu) Estudos de Sarcoma de Ewing. Journal of clinical oncology: revista oficial da Sociedade Americana de Oncologia Clínica 1998; 16: 3044-3052.
- Letourneau PA, et al: A ressecção de metástases pulmonares em doentes pediátricos com sarcoma de Ewing melhora a sobrevivência. Journal of pediatric surgery 2011; 46: 332-335.
- Ladenstein R, et al: Sarcoma de Ewing multifocal disseminado em primeiro lugar: resultados do ensaio Euro-EWING 99. Journal of clinical oncology: revista oficial da Sociedade Americana de Oncologia Clínica 2010; 28: 3284-3291.
- Dunst J, Paulussen M, Jurgens H: Irradiação pulmonar para sarcoma de Ewing com metástases pulmonares no diagnóstico: resultados dos estudos CESS. Radioterapia e Oncologia: Órgão da Sociedade Alemã Rontgen [et al] 1993; 169: 621-623.
- Paulussen M, et al.: Primary metastatic (stage IV) Ewing tumor: análise de sobrevivência de 171 pacientes dos estudos EICESS. Estudos de Sarcoma de Ewing Cooperativa Europeia Intergrupal. Anais de oncologia: revista oficial da Sociedade Europeia de Oncologia Médica/ESMO 1998; 9: 275-281.
- Oberlin O, et al.: Impact of high-dose busulfan plus melphalan as consolidation in metastatic Ewing tumors: a study by the Societe Francaise des Cancers de l’Enfant. Journal of clinical oncology: revista oficial da Sociedade Americana de Oncologia Clínica 2006; 24: 3997-4002.
- Ladenstein R, et al: Impacto da megaterapia em crianças com tumores de Ewing de alto risco em completa remissão: um relatório do Registo de Tumores Sólidos EBMT. Transplante de medula óssea 1995; 15: 697-705.
- Dirksen U, et al.: Eficácia da consolidação da quimioterapia de alta dose de busulfan-melphalan (BuMel) em comparação com a quimioterapia convencional combinada com a irradiação pulmonar no sarcoma de ewing (ES) com metástases pulmonares primárias: Resultados do ensaio aleatório EURO-EWING 99-R2pulm (EE99R2pul). Journal of Clinical Oncology 2016; 34(15) Suplemento: 11001-11001.
- Weston CL, et al: Estabelecer a sobrevivência a longo prazo e a cura em pacientes jovens com sarcoma de Ewing. Revista britânica sobre o cancro 2004; 91: 225-232.
- Stahl M, et al: Risco de recidiva e sobrevivência após recaída em doentes com sarcoma de Ewing. Sangue pediátrico e cancro 2011; 57: 549-553.
- Bacci G, et al: Ressecção de metástases pulmonares metacrónicas em doentes com sarcoma de Ewing inicialmente tratados com quimioterapia adjuvante ou neoadjuvante. Eur J Cancer 1995; 31A: 999-1001.
- Rasper M, et al: O valor da quimioterapia de alta dose em pacientes com o primeiro sarcoma de Ewing recidivado. Sangue pediátrico e cancro 2014; 61: 1382-1386.
- Hunold A, et al: Topotecan e ciclofosfamida em doentes com tumores de Ewing refractários ou recidivados. Sangue pediátrico e cancro 2006; 47: 795-800.
- van Maldegem AM, et al: Etoposide e terapia combinada de carbo-ou cisplatina em sarcoma de Ewing refractário ou recidivado: um grande estudo retrospectivo. Sangue pediátrico e cancro 2015; 62: 40-44.
- Casey DA, et al: Irinotecan e temozolomide para o sarcoma de Ewing: a experiência do Memorial Sloan-Kettering. Sangue pediátrico e cancro 2009; 53: 1029-1034.
- Ginsberg JP, et al: Long-term survivors of childhood Ewing sarcoma: relatório do estudo dos sobreviventes do cancro infantil. Journal of the National Cancer Institute 2010; 102: 1272-1283.
InFo ONCOLOGY & HEMATOLOGY 2018; 6(5): 13-16.