O foco da terapia ainda está nas opções de tratamento sintomático e na prevenção de factores de provocação. As opções terapêuticas moleculares, potencialmente também curativas para a epidermólise bolhosa ainda se encontram na fase de investigação. No trabalho de diagnóstico desta doença auto-imune geno- e fenotípica heterogénea, há uma série de coisas que precisam de ser consideradas.
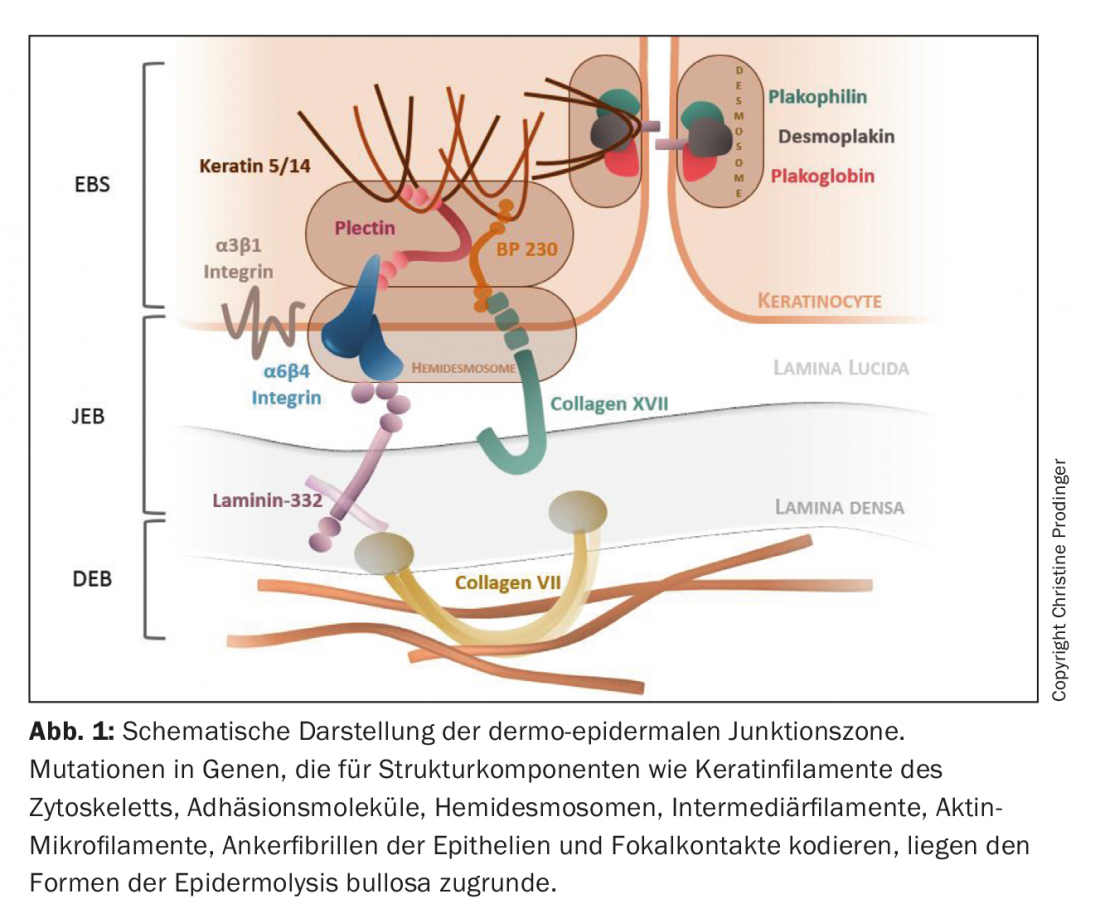
A Epidermólise Bulhosa (EB) é um grupo de geno- e fenotípicos genodermatoses heterogéneos que se caracterizam por uma excessiva fragilidade mecânica dos tecidos epitelizados. Com uma prevalência de cerca de 500.000 casos em todo o mundo, a EB é uma doença rara [1,2]. Até à data, as mutações têm sido descritas em mais de 20 genes que codificam componentes envolvidos na montagem dos filamentos de queratina do citoesqueleto, moléculas de adesão, desmosmosomas, hemidesmosomas e fibrilas de ancoragem de epitélios (Fig. 1). Consequentemente, a integridade estrutural e funcional da adesão intra-epidérmica e da aderência dermoepidérmica à pele e às membranas mucosas é prejudicada, afectando a função de barreira, a interacção célula-célula e célula-matriz, a proliferação, a regeneração dos tecidos e os processos de diferenciação [3–5]. O espectro combinatório de tipo, homo- ou heterozigosidade, número (herança mono ou digenética) e localização da mutação no respectivo segmento genético, bem como a resultante perturbação quantitativa (ausência ou redução) e qualitativa (perda gradual da função) da expressão proteica, causa consideráveis variações geno- e consequentemente fenotípicas da EB. Para além do defeito genético primário, factores secundários epigenéticos e bioquímicos (por exemplo, indução de cascatas inflamatórias crónicas e remodelação de tecidos), bem como factores ambientais, também influenciam a gravidade clínica [6,7].
Uma vez que os genes índice afectados não são apenas expressos na pele e membranas mucosas próximas da pele, mas também noutros epitélios (vias respiratórias, urogenitais e gastrintestinais) e no tecido mesenquimal (músculos esqueléticos), também aí são possíveis manifestações extracutâneas primárias. A EB pode assim evoluir para uma doença sistémica com uma morbilidade e mortalidade significativas [8].
Classificação fisiopatológica
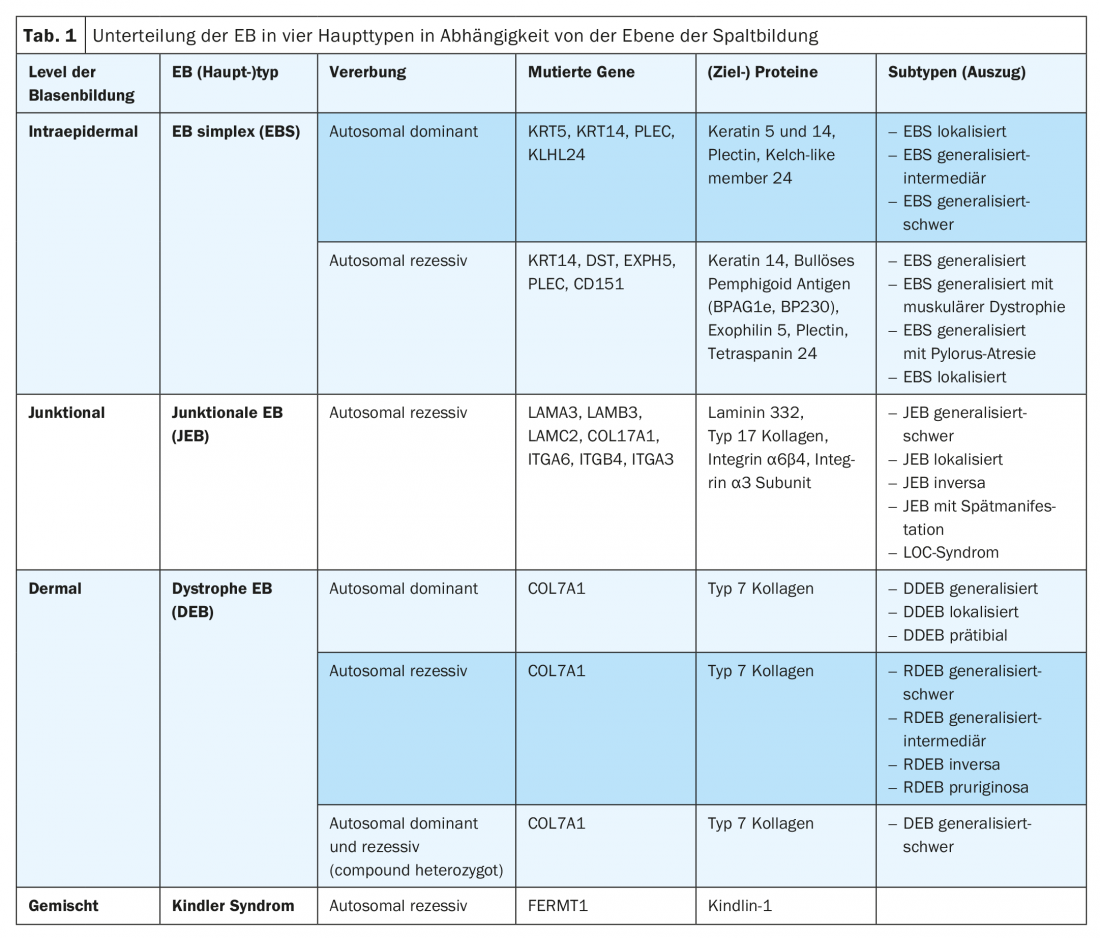
O principal sintoma da EB é o aumento da fragilidade excessiva da pele e das mucosas contra o stress mecânico com a formação de bolhas, erosões, ulcerações, crostas ou cicatrizes, dependendo do subtipo. De acordo com o nível de clivagem como função e consequência da mutação subjacente, a EB está dividida em quatro tipos principais (Tab. 1) [9]. Devido à crescente disponibilidade de métodos modernos de diagnóstico molecular (por exemplo, “sequenciação da próxima geração”), novos (sub)tipos de EB estão também a ser descritos uma e outra vez. Assim, uma mutação no gene KLHL24 que codifica um componente do complexo da ligase ubiquitina poderia ser atribuída a uma variante autossómica dominante do simplex EB, o que resulta numa ubiquitinação excessiva e na degradação da queratina 14 [10–12]. Num fenótipo semelhante ao de Kindler, foi também recentemente identificada uma mutação no gene CD151, que codifica uma tetraspanina na zona da membrana da cave. Esta proteína transmembrana interage com as integrinas e está envolvida em processos de crescimento, desenvolvimento e motilidade celular [13]. Além disso, foi recentemente descoberta uma mutação no gene PLOD3, que codifica a lisil hidroxilase 3 e regula o processamento pós-traducional do colagénio tipo 7. Clinicamente, os indivíduos afectados apresentam extensos defeitos do tecido conjuntivo, contraturas articulares, malformações do esqueleto e retardamento do crescimento. O nível de formação de bolhas é semelhante ao da EB distrófica recessiva na densa sublamina [14].
Algoritmo de diagnóstico
Especialmente no recém-nascido com formação de bolhas, uma génese traumática, metabólica, hematológica, infecciosa, medicinal e auto-imune deve ser excluída em primeiro lugar [15]. Subsequentemente, é aplicado um algoritmo de diagnóstico:
- (Família) história e clínica
- Determinação da contaminação microbiana (por exemplo, esfregaços, PCR, serologia)
- Biópsia perilesional para avaliação histológica de possíveis diagnósticos diferenciais
- Imunofluorescência directa paralela para determinar o nível de clivagem e expressão semi-quantitativa da proteína a partir de uma borbulha induzida (por exemplo, rotação de um apagador de lápis até ao desenvolvimento do eritema) e bolha fresca (não superior a 12 horas) na pele não exposta ao sol.
- Microscopia electrónica de transmissão para determinar o plano de clivagem e defeitos morfológicos
- Análise da mutação (dependente do genoma inteiro, exoma, cluster, sequenciação de painéis)
O (sub)tipo de EB respectivo pode ser determinado de acordo com os resultados recolhidos. O plano de clivagem determinado por microscopia electrónica de transmissão e imunofluorescência define o principal tipo de EB. O fenótipo clínico é definido através da indicação da gravidade relativa (suave, intermédia, grave) e do padrão de distribuição (localizada, generalizada) – quando apropriado, são também indicados sintomas característicos, tais como pseudosinndactilia. Para além de resultados específicos recolhidos em microscopia electrónica de transmissão ou mapeamento de imunofluorescência, são listadas as proteínas e genes específicos afectados, o tipo de mutação ou mutação específica [9]. Os sucessos na caracterização molecular da base patogénica dos diferentes tipos de EB também permitem um aconselhamento genético mais preciso, prognóstico e diagnóstico pré-natal e são o pré-requisito básico para intervenções terapêuticas inovadoras e orientadas.
Princípios terapêuticos
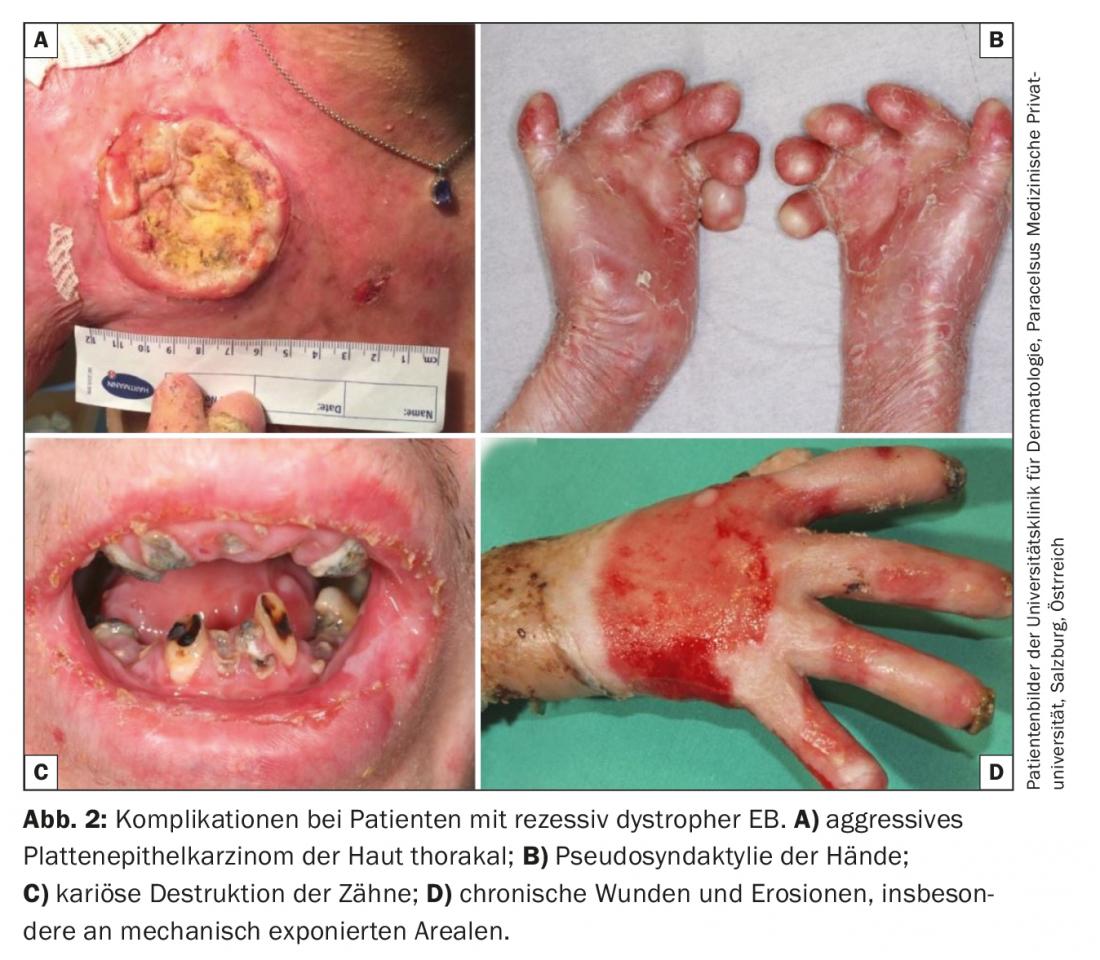
Na ausência de opções de tratamento curativo na prática clínica, o foco da terapia para todas as formas de EB está em evitar factores de provocação e optimizar as abordagens terapêuticas sintomáticas . Estes incluem terapia local adequada à fase da ferida, melhoria ou manutenção da barreira cutânea através de cuidados de pele optimizados, alívio da dor e comichão, prevenção e tratamento de infecções (terapia antisséptica e antibiótica intermitente), manutenção de ingestão adequada de calorias e nutrientes, e finalmente prevenção e tratamento de complicações tais como anemia, osteoporose e carcinoma espinocelular agressivo. (Fig. 2) [16,17].
As perspectivas de opções de terapia molecular, potencialmente também curativa para a EB são, em princípio, encorajadoras, mesmo que a sua implementação ampla, segura, (sustentável) eficiente e praticável na prática clínica diária dos pacientes com EB ainda seja difícil de avaliar actualmente. O espectro dos métodos é complexo:
Terapia génica: Em 2006, a terapia génica utilizando o transplante de queratinócitos de correcção genética LAMB3 foi realizada pela primeira vez num doente com JEB (mutação no gene LAMB3) autossómico recessivo [18]. Nesta terapia de “substituição genética”, células cutâneas cultivadas foram transfectadas com um vector contendo uma cópia cDNA funcional do gene mutante causador e transplantadas para áreas da ferida como equivalentes epiteliais da pele. A zona de junção dermo-epidérmica tem sido estrutural e funcionalmente estável durante o período de seguimento de 14 anos até à data, sem evidência de bolhas, inflamação ou reacção imunitária ao neoantigénio introduzido terapeuticamente. Num estudo recente, foram cultivados queratinócitos autólogos segundo o mesmo princípio, as células estaminais epidérmicas foram isoladas e especificamente corrigidas pelo gene, expandidas e depois transplantadas para grandes áreas de feridas de um rapaz de 7 anos com JEB (mutação no gene da lamina 332). Assim, uma reepitelização até 80% da superfície do corpo poderia ser alcançada. A resposta clínica correlacionada com a expressão persistente da lamina 332 até à data [19]. A eficiência é limitada pelo (também) pequeno número de células estaminais epidérmicas nas culturas primárias de pacientes com EB para assegurar a correcção permanente, que se estão a esgotar devido a feridas crónicas e ao avanço da idade, bem como aspectos de segurança (no que diz respeito a vectores virais e genotoxicidade ou mutagénese induzida por inserção). Os desenvolvimentos tecnológicos estão a ser avaliados em estudos actuais e devem melhorar o perfil de risco-benefício deste método, que é particularmente adequado para feridas crónicas circunscritas altamente sintomáticas ou em risco de complicações a longo prazo [20].
O “silenciamento de genes” para formas autossómicas dominantes de EB (ou seja, silenciar o alelo mutante com “pequenos RNAs interferentes”), bem como abordagens de terapia genética de aplicação fácil e tópica estão também em desenvolvimento. Neste último caso, por exemplo, o vesículas extracelulares, que são isoladas de células estaminais mesenquimais alogénicas e podem transportar tanto a proteína de colagénio de tipo 7 em falta como o mRNA COL7A1 para células-alvo [21, 22].
As tecnologias de edição de genoma que utilizam propriedades de nucleases programáveis (por exemplo CRISPR/Cas9, TALEN, nucleases de dedos de zinco) são baseadas na modificação/correcção de sequências de genes mutantes. Neste processo, é especificamente induzida uma quebra dupla no ADN, as sequências mutantes são eliminadas e as sequências estranhas correctivas são inseridas, ou as bases individuais são corrigidas por mecanismos de replicação celular. No entanto, as preocupações de segurança, especialmente no que diz respeito à precisão insuficiente e aos potenciais sítios de ligação fora do alvo, ainda não justificam a utilização (in vivo) em humanos [23,24].
Um fenómeno clínico notável que pode ocorrer em todas as principais formas de EB é a ocorrência (espontânea) de áreas de pele saudável sem bolhas como resultado de eventos genéticos correctivos, o que é chamado mosaicismo revertente ou “terapia genética natural”. As tentativas de transplantar queratinócitos reversíveis obtidos em ensaios clínicos de fase I, utilizando biópsias perfuradas em feridas de EB afectadas após expansão in vitro, ficaram, até agora, aquém das expectativas, principalmente devido à perda progressiva de células reversíveis [25,26].
Terapia celular: Os primeiros resultados do transplante de células estaminais de medula óssea mostraram células doadoras na pele (presumivelmente células estaminais pluripotentes de medula óssea reprogramadas) e boa resposta, em parte ao longo de vários anos, em alguns pacientes com EB distrófica recessiva, apesar da falta de provas de uma concentração de colagénio do tipo 7 restituída. Este sucesso, cujo mecanismo subjacente ainda não é claro, foi diminuído por um aumento significativo da mortalidade periprocedural. As terapias de condicionamento e os protocolos de transplante revistos devem melhorar a tolerância [27,28].
Os substratos das terapias celulares alternativas incluem células estaminais mesenquimais alogénicas aplicadas por via intradérmica ou intravenosa e células estaminais mesenquimais adipogénicas. Foi possível observar, pelo menos temporariamente, uma melhor cicatrização das feridas e uma redução dos sinais inflamatórios na pele, o que provavelmente se deve principalmente à indução de processos imunomoduladores favoráveis [29]. Já foi demonstrado um efeito clínico em estudos iniciais com fibroblastos autólogos de tipo selvagem injectados por via intradérmica ou de tipo genético, que também produzem colagénio de tipo 7, para além de queratinócitos [30,31].
Células semelhantes às células estaminais embrionárias pluripotentes também podem ser derivadas de células somáticas (por exemplo, fibroblastos ou queratinócitos) por transfecção de três a quatro factores de transcrição embrionária. A utilização destas chamadas células estaminais pluripotentes induzidas (iPSC), que podem novamente diferenciar-se em vários tipos de células (por exemplo queratinócitos ou fibroblastos), está também a ser investigada na EB em estudos pré-clínicos. A utilização de células reversíveis/keratinócitos para a produção de iPSC tem grande potencial, uma vez que a correcção genética e os riscos associados podem ser dispensados com [32,33].
Terapias proteicas: Também se tenta corrigir a zona de junção dermo-epidérmica defeituosa, substituindo a proteína em falta ou produzida de forma defeituosa. Enquanto os estudos com aplicação de colagénio recombinante tipo 7 ainda se encontram na fase pré-clínica, a aplicação tópica de um gel contendo um tipo modificado 7 herpes simplex tipo 1, por exemplo, já está a ser testado em ensaios clínicos [34,35].
Terapias baseadas em RNA/”pequenas moléculas”: As mutações disparatadas, que causam um códão de paragem através de uma mutação de ponto de ADN e assim abortam a tradução, são responsáveis por cerca de 10% de todas as doenças genéticas humanas. Drogas como os aminoglicosídeos (por exemplo, gentamicina) ou o imunomodulador amlexanox podem levar a uma “leitura” do códão de paragem (por exemplo, a mutação COL7A1), ligando-se aos ribossomas na presença desta mutação, permitindo assim a produção de proteínas funcionais [36].
As modificações a nível do ARN são também conseguidas por meio de “pulo de exão oligonucleótido antisense” (remoção direccionada de exões contendo mutações) e “emendas de emendas de RNA mediadas por emendas” (SMaRT) (correcção de segmentos de pré-RNA mutantes). Esta última tecnologia já foi utilizada pré-clinicamente para corrigir com sucesso uma mutação do gene plectin em EB simplex e uma mutação COL7A1 em EB distrófica recessiva, bem como mutações autossómicas dominantes no gene queratino-14 de uma linha de células EB simplex [37,38].
As chamadas “pequenas moléculas” são utilizadas como mediadores de um efeito “modificador da doença”. Estes incluem o calcipotriol tópico, que se pensa impulsionar as defesas antimicrobianas endógenas e melhorar a cicatrização de feridas aumentando a expressão do peptídeo antimicrobiano cathelicidina [39] e a diacereína tópica, um componente da raiz do ruibarbo e um potente inibidor do pró-inflamatório IL-1β, que demonstrou reduzir a formação de bolhas em doentes com SEE de acordo com dados preliminares publicados. [40]. O rigosertib inibidor da tirosina quinase oral também mostra inibição selectiva das células escamosas do tumor de células de doentes com EB distrófica recessiva in vitro, e a sua eficácia e segurança estão a ser testadas num estudo em curso. O carcinoma espinocelular altamente agressivo como complicação da EB crónica As feridas são uma das principais causas de morte, particularmente em doentes com EB atrófica recessiva [41]. Para além do rigosertib, o anticorpo monoclonal receptor anti-PD-1 nivolumab está actualmente a ser submetido a testes controlados quanto à sua eficácia no carcinoma escamoso escamoso localmente avançado e metastático na coorte EB (EudraCT 2016-002811-16).
Mensagens Take-Home
- A Epidermólise Bulhosa (EB) é uma doença geno- e fenotípica heterogénea. Os cursos severos transformam-se numa doença multi-sistemas com morbilidade e mortalidade pronunciadas. As principais causas de morte são infecções, distrofia, falência de órgãos e carcinoma de células escamosas. Estas últimas ocorrem cedo e multiplicam-se em feridas crónicas e mostram um curso agressivo.
- O diagnóstico é feito em correlação de clínica, histologia, imunofluorescência e análise molecular. Apesar das abordagens inovadoras, parcialmente causais das estratégias de terapia molecular, a cura é ainda uma perspectiva futura indirectamente vaga. A imunomodulação, por sua vez, é uma estratégia de tratamento promissora.
- Os componentes secundários epigenéticos e bioquímicos, bem como os factores ambientais, que podem induzir, por exemplo, cascatas inflamatórias crónicas e sistemicamente eficazes, têm uma elevada relevância patogénica. Tal como acontece com outras doenças raras, certas características dificultam a realização de ensaios clínicos e, por conseguinte, geram provas de alta qualidade.
Literatura:
- Fine JD: epidermólise bullosa hereditária. Orphanet J Rare Dis 2010; 5: 12.
- Fine JD, et al: Epidermolysis bullosa and the risk of life-threatening cancers: the National EB Registry experience, 1986-2006. J Am Acad Dermatol 2009; 60(2): 203-211.
- Fine JD, et al: A classificação da epidermólise bullosa hereditária (EB): Relatório da Terceira Reunião de Consenso Internacional sobre Diagnóstico e Classificação da EB. J Am Acad Dermatol 2008; 58(6): 931-950.
- Bruckner-Tuderman L, et al: Progress in epidermolysis bullosa research: summary of DEBRA International Research Conference 2012. J Invest Dermatol 2013; 133(9): 2121-2126.
- Uitto J, Richard G: Progresso na epidermólise bolhosa: classificação genética e implicações clínicas. Am J Med Genet C Semin Med Genet 2004; 131C(1): 61-74.
- Kuttner V, et al.: Remodelação global do microambiente celular devido à perda de colagénio VII. Mol Syst Biol 2013; 9: 657.
- Odorisio T, et al.: Gémeos monozigóticos discordantes para o fenótipo da epidermólise bolhosa distrófica recessiva realçam o papel da sinalização TGF-beta na modificação da gravidade da doença. Hum Mol Genet 2014; 23(15): 3907-3922.
- Pulkkinen L, et al: Novel ITGB4 mutações em variantes letais e não letais de epidermólise bolhosa com atresia pilórica: missense versus nonsense. Am J Hum Genet 1998; 63(5): 1376-1387.
- Fine JD, et al: Inherited epidermolysis bullosa: recomendações actualizadas sobre diagnóstico e classificação. J Am Acad Dermatol 2014; 70(6): 1103-1126.
- He Y, et al: Mutações monoalleicas no códão de iniciação de tradução do KLHL24 causam fragilidade da pele. Am J Hum Genet 2016; 99(6): 1395-1404.
- Lee JYW, et al: Mutations in KLHL24 Add to the Molecular Heterogeneity of Epidermolysis Bullosa Simplex. J Invest Dermatol 2017; 137(6): 1378-1380.
- Lin Z, et al.: As mutações estabilizadoras da ubiquitina ligase KLHL24 causam perda de queratina 14 e fragilidade da pele humana. Nat Genet 2016; 48(12): 1508-1516.
- Vahidnezhad H, et al: Mutação recessiva em tetraspanina CD151 causa epidermólise bolhosa tipo síndrome de Kindler com manifestações multi-sistémicas incluindo nefropatia. Matrix Biol 2018; 66: 22-33.
- Salo AM, et al: Uma perturbação do tecido conjuntivo causada por mutações do gene da lisil hidroxilase 3. Am J Hum Genet 2008; 83(4): 495-503.
- Nischler E, et al: Diagnostic pitfalls em recém-nascidos e bebés com bolhas e erosões. Dermatol Res Pract 2009; 2009: 320403.
- El Hachem M, et al: Recomendações de consenso multicêntrico para cuidados de pele na epidermólise bolhosa hereditária. Orphanet J Rare Dis 2014; 9: 76.
- Dănescu S, et al: Correlação entre gravidade da doença e qualidade de vida em doentes com epidermólise bolhosa. J Eur Acad Dermatol Venereol 2018. doi: 10.1111/jdv.15371. [Epub ahead of print]
- Mavilio F, et al: Correcção da epidermólise bullosa juncional por transplante de células estaminais epidérmicas geneticamente modificadas. Nat Med 2006; 12(12): 1397-1402.
- Hirsch T, et al.: Regeneração de toda a epiderme humana utilizando células estaminais transgénicas. Natureza, 2017; 551(7680): 327-332.
- Siprashvili Z, et al: Segurança e resultados de feridas após enxertos epidérmicos autólogos geneticamente corrigidos em doentes com Epidermólise Distroférica Bullosa Recessiva. JAMA 2016; 316(17): 1808-1817.
- Rosa J, et al: Current Non-viral SiRNA Delivery Systems as a Promising Treatment of Skin Diseases. Curr Pharm Des 2018; 24(23): 2644-2663.
- McBride JD, et al: Mecanismo duplo de transferência de colagénio do tipo VII por vesículas extracelulares de células estaminais mesenquimais da medula óssea para fibroblastos epidermolise bolhosa distrófica recessiva. Biochimie 2018; 155: 50-58.
- Hainzl S, et al: COL7A1 Edição via CRISPR/Cas9 em Epidermólise Distrófica Recessiva Bullosa. Mol Ther 2017; 25(11): 2573-2584.
- March OP, Reichelt J, Koller U: edição de genes para doenças de pele: nucleases de desenhadores como ferramentas para terapia genética de doenças de fragilidade da pele. Exp Physiol 2018; 103(4): 449-455.
- Gostynski A, Pasmooij AM, Jonkman MF: Transplante terapêutico bem sucedido de pele reversa em epidermólise bolhosa. J Am Acad Dermatol 2014; 70(1): 98-101.
- van den Akker PC, et al: Uma “célula revertente de late-but-fitter” explica a alta frequência do mosaicismo revertente na epidermólise bolhosa. PLoS One 2018; 13(2): p. e0192994.
- Ebens CL, et al: Transplante de medula óssea com ciclofosfamida pós-transplante para a epidermólise bolhosa distrófica recessiva expande o conjunto de dadores relacionados e permite a tolerância de enxertos celulares não-hematopoiéticos. Br J Dermatol 2019. doi: 10.1111/bjd.17858. [Epub ahead of print]
- Vanden Oever M, et al: Inside out: medicina regenerativa para a epidermólise bolhosa distrófica recessiva. Pediatr Res 2018; 83(1-2): 318-324.
- Ganier C, et al: Injeção intradérmica de células estromais mesenquimais de medula óssea corrige a Epidermólise Distrófica Recessiva Bullosa num modelo de Xenograft. J Invest Dermatol 2018; 138(11): 2483-2486.
- Petrof G, et al: A terapia com células fibroblásticas melhora a cicatrização inicial em feridas bolhosas de epidermólise distrófica recessiva: resultados de um ensaio aleatório, controlado por veículo. Br J Dermatol 2013; 169(5): 1025-1033.
- Venugopal SS, et al: Um ensaio fase II aleatório controlado por veículo de fibroblastos alogénicos intradérmicos para a epidermólise bolhosa distrófica recessiva. J Am Acad Dermatol 2013; 69(6): 898-908.e7. doi: 10.1016/j.jaad.2013.08.014
- Itoh M, Kiuru M, Cairo MS, Christiano AM.: Geração de queratinócitos a partir de células estaminais pluripotentes normais e recessivas da epidermólise distrófica induzida por bullosa-induzida. Proc Natl Acad Scien U S A 2011; 108(21): 8797-8802.
- Nakayama C, et al: O desenvolvimento de células estaminais pluripotentes induzidas derivadas de células estaminais mesenquimais a partir de queratinócitos humanos normais e queratinócitos epidérmicos RDEB. J Dermatol Sci 2018; 91(3): 301-310.
- South AP, Uitto J: Terapia de Substituição de Colagénio Tipo VII em Epidermólise Distrófica Recessiva Bullosa-Quanta, Quantas vezes? J Invest Dermatol 2016; 136(6): 1079-1081.
- NIH: Topical Bercolagene Telserpavec (KB103) Gene Therapy to Restore Functional Collagen VII for the Treatment of Dystrophic Epidermolysis Bullosa (GEM-1), https://clinicaltrials.gov/ct2/show/NCT03536143, último acesso 25 Mar 2019.
- Lincoln V, et al: A gentamicina induz a leitura sem sentido da mutação LAMB3 e restaura a lamina funcional 332 na epidermólise bullosa juncional. Proc Natl Acad Sci U S A, 2018; 115(28): E6536-E6545.
- Pequim P, et al: A Gene Gun-mediated Nonviral RNA trans-splicing Strategy for Col7a1 Repair. Mol Ther Ácidos Nucleicos 2016. 5:e287. doi: 10.1038/mtna.2016.3.
- Turczynski S, et al: Targeted Exon Skipping Restaura a Expressão de Colagénio Tipo VII e Formação de Fibrilha Ancoragem num Modelo In Vivo RDEB. J Invest Dermatol 2016; 136(12): 2387-2395.
- Guttmann-Gruber C, et al.: A baixa dose de calcipotriol pode provocar o fecho de feridas, efeitos antimicrobianos e anti-neoplásicos em queratinócitos de epidermólise bolhosa. Rep. Sci 2018; 8(1): 13430.
- Wally V, et al: Diacerein orphan drug development for epidermolysis bullosa simplex: Um ensaio clínico fase 2/3 randomizado, controlado por placebo, duplo-cego. J Am Acad Dermatol 2018; 78(5): 892-901.e7. doi: 10.1016/j.jaad.2018.01.019.
- Atanasova VS, et al: Identificação de rigosertib para o tratamento do carcinoma de células escamosas associadas à epidermólise distrófica recessiva bullosa-cármaco escamoso. Clin Cancer Res, 2019. doi: 10.1158/1078–0432.CCR-18–2661
PRÁTICA DA DERMATOLOGIA 2019; 29(2): 16-20