O termo colectivo “hipertensão pulmonar (HP)” abrange vários tipos de hipertensão pulmonar com risco de vida, cujas abordagens terapêuticas são muito diferentes e altamente complexas. Uma vez que os sintomas são frequentemente pouco específicos, mesmo o diagnóstico é um grande desafio.
A classificação clínica da hipertensão pulmonar (PH) está actualmente dividida em cinco grupos, como o Prof. Dr. Horst Olschewski, Chefe do Departamento Clínico de Pneumologia, Departamento Universitário de Medicina Interna, Hospital Universitário LKH Graz, nos lembrou no início:
- O grupo 1 inclui a Hipertensão Arterial Pulmonar (HAP), a forma mais rara de hipertensão pulmonar, que só é diagnosticada depois de outras causas subjacentes terem sido descartadas.
- O grupo 2, hipertensão pulmonar na doença cardíaca esquerda, é de longe a forma mais comum de hipertensão pulmonar.
- O grupo 3 inclui a hipertensão pulmonar devido a doença pulmonar e/ou deficiência de oxigénio. Esta forma também ocorre com muito mais frequência do que a HAP.
- O grupo 4 inclui a hipertensão pulmonar após embolia pulmonar, que afecta até 4% dos pacientes que sofreram uma embolia pulmonar aguda.
- O grupo 5 inclui formas raras de hipertensão pulmonar que não podem ser claramente atribuídas a nenhum dos grupos acima mencionados, tais como a hipertensão pulmonar em sarcoidose ou doença renal crónica.
Os grupos 2 e 3, de acordo com o Prof. Olschewski, representam em conjunto um total de 90% de toda a hipertensão pulmonar, o que apresenta perigos de diagnóstico diferencial.
Heterogeneidade do IPAH
Para análise de agregados, foram identificados os pacientes que preenchiam os critérios para hipertensão arterial pulmonar idiopática (IPAH).
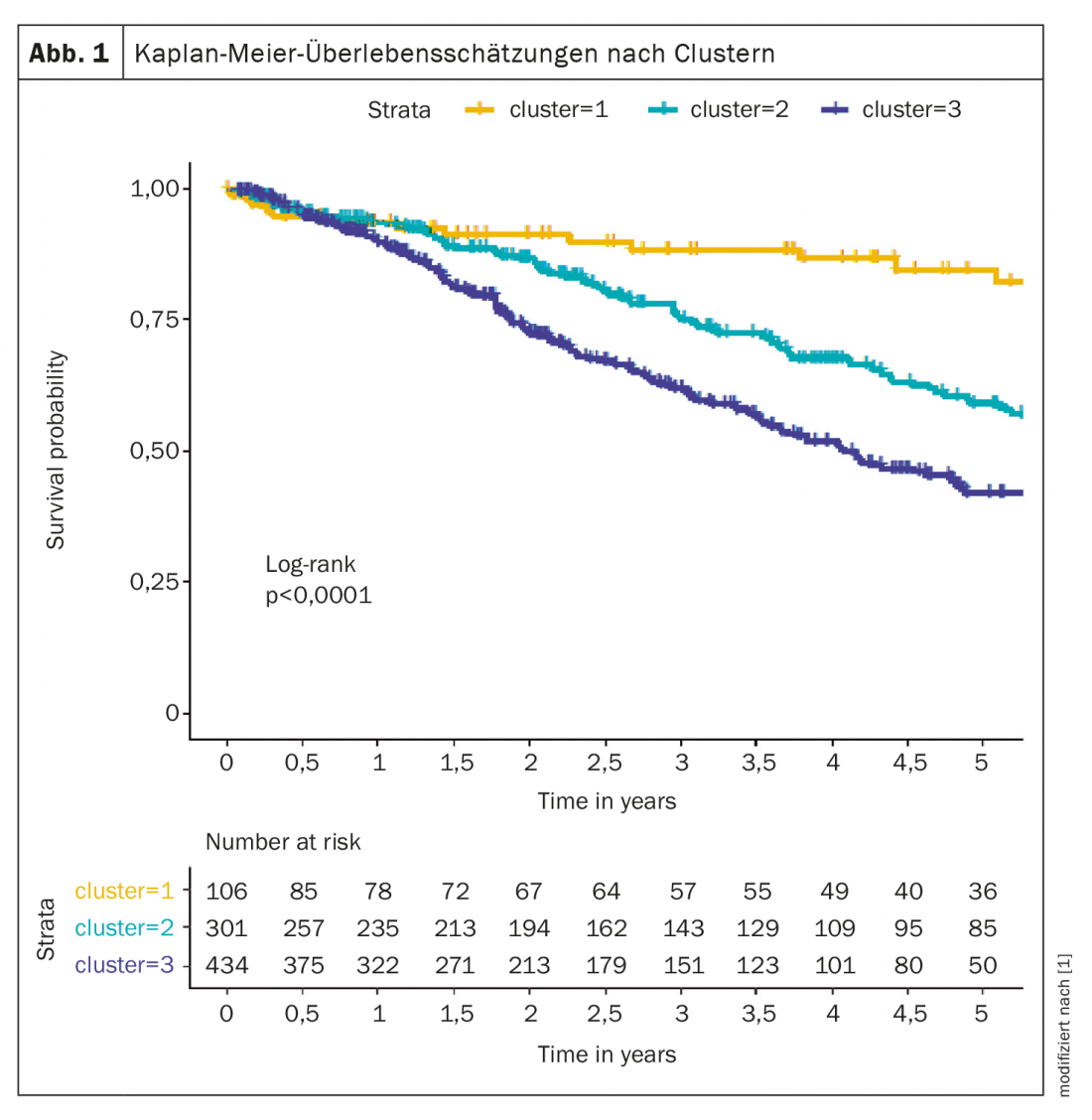
Por exemplo, uma análise de agrupamento identificou diferentes fenótipos que diferem na apresentação clínica, resposta à terapia e sobrevivência. Os grupos analisados foram compostos a partir do registo do COMPERA [1], foram definidos três grupos: grupo 1 (n=106; 12,6%) incluía uma idade média de 45 anos, 76% eram mulheres, sem comorbilidades, na maioria nunca fumadores, DLCO ≥45%; grupo 2 (n=301; 35,8%) incluía pacientes cuja idade média era de 75 anos, 98% mulheres, comorbilidades frequentes, historial de não fumadores, DLCO na maioria ≥45%; grupo 3 (n=434; 51,6%) consistia em indivíduos com uma idade média de 72 anos, 72% eram homens, comorbidades frequentes, historial de tabagismo e DLCO na sua maioria abaixo de 45%.
Os doentes do grupo 1 responderam melhor ao tratamento de HAP do que os doentes dos outros dois grupos. A taxa de sobrevivência de cinco anos foi de 84,6% no agregado 1, 59,2% no agregado 2 e 42,2% no agregado 3 (Fig. 1). Segundo o perito, estes dados sugerem que são necessários critérios para distinguir pacientes atípicos do IPAH dos verdadeiros pacientes do IPAH.
Diagnóstico diferencial por vezes difícil
Como as consequências terapêuticas dependem em grande medida da causa da hipertensão pulmonar, é importante completar os procedimentos de diagnóstico e determinar a causa principal da PH antes de tomar uma decisão sobre a medicação para a HAP. O World Symposia on Pulmonary Hypertension (WSPH) produziu directrizes para estas importantes decisões. PH do grupo 2 ou doenças de desenvolvimento complexas com aumento da pressão pós-capilar podem ser reconhecidas com relativa facilidade pelo aumento da pressão da cunha arterial pulmonar. PH do grupo 4 pode ser detectado ou excluído através de varreduras pulmonares de perfusão em combinação com uma TAC ao tórax. Grupo 1 PAH e Grupo 3 PH são perfis de doença bastante diferentes mas por vezes podem ser difíceis de distinguir. A WSPH sugere que a hipertensão pulmonar grave combinada com um ligeiro comprometimento no teste de função pulmonar (FEV1 >60 e FVC >60%), anomalias parenquimatosas ligeiras na TC de alta resolução do tórax e comprometimento circulatório no teste de exercício cardiopulmonar são indicativos do grupo HAP 1. Estes pacientes são candidatos à terapia de HAP. Se o doente sofrer de PH do grupo 3, a única indicação possível para a terapia de HAP é hipertensão pulmonar grave (mPAP ≥35 mmHg ou mPAP entre 25 e 35 mmHg juntamente com um índice cardíaco (IC) muito baixo <2.0 L/min/m2), que só pode ser derivada invasivamente, disse o Prof Olschewski.
Um estudo também analisou a diferenciação entre pacientes com HAP (grupo 1) e pacientes com insuficiência cardíaca (grupo 2) [2]. Embora o aumento da pressão de enchimento do lado esquerdo e a regurgitação mitral funcional sejam as causas primárias da PH pós-capilar, as directrizes e recomendações diferenciam entre a PH pós-capilar isolada (IpcPH) e a PH pós e pré-capilar combinada (CpcPH). Esta última é definida pela resistência vascular pulmonar (PVR) aumentada para unidades de Madeira (WE). É importante diferenciar entre a definição geral de PH (mPAP >20 mmHg) e a definição de PH pré-capilar incluindo a HAP, para a qual é necessária uma pressão de cunha arterial pulmonar (PAWP) ≤15 mmHg e um aumento da resistência vascular pulmonar (PVR) para ≥3 Unidades de madeira também é necessária. De acordo com isto, a terapia medicamentosa orientada para HAP é indicada para um PAWP ≤15 mmHg.
A diferenciação dos pacientes com HAP e DPOC (grupo 1) dos pacientes com PH devido à DPOC (grupo 3) também não é fácil. A maioria dos pacientes com DPOC com PH pertencem ao grupo 3. Alguns pacientes com DPOC com PH e aumento da pressão de enchimento ventricular esquerdo (PH pós-capilar) causado por doença cardiovascular concomitante são atribuídos ao grupo 2. As doenças tromboembólicas crónicas também podem causar PH, especialmente porque a DPOC é um factor de risco para o tromboembolismo venoso, estes doentes com DPOC são geralmente atribuídos ao grupo 4. Pensa-se que os doentes com obstrução muito ligeira das vias aéreas periféricas e PH pré-capilar grave com grande aumento da resistência vascular pulmonar (PVR) e baixo débito cardíaco (CO) têm predominantemente HAP (grupo 1) com DPOC ligeira como condição associada. Na maioria dos casos, o PH é relativamente ligeiro em pacientes com DPOC, explicou o pneumologista, mas num subconjunto de pacientes com DPOC, a presença de certas características clínicas sugere um “fenótipo vascular pulmonar”. Tal fenótipo seria caracterizado por um PH pré-capilar grave com resistência vascular pulmonar acentuadamente aumentada, limitação moderada do fluxo de ar, capacidade difusora gravemente reduzida de monóxido de carbono, normo- ou hipocapnia, limitação da carga circulatória e insuficiência cardíaca direita progressiva.
Outro estudo tentou determinar limiares hemodinâmicos prognosticamente relevantes para o PH grave na DPOC, utilizando uma abordagem imparcial [3]. A PVR >5 WU foi considerado o mais forte preditor independente de hemodinâmica da mortalidade em pacientes com DPOC. Este limiar é o melhor para identificar pacientes com DPOC com doença vascular pulmonar grave.
Avanços na terapia medicamentosa
Os recentes avanços na terapia medicamentosa para HAP não se devem à descoberta de novos caminhos de sinalização, mas ao desenvolvimento de novas estratégias de terapia combinada e à escalada de tratamentos baseados na avaliação sistemática da resposta clínica. A estratégia de tratamento baseia-se na gravidade do paciente com HAP recentemente diagnosticado, que é determinada utilizando uma abordagem multiparamétrica de estratificação de risco. Os parâmetros clínicos, de exercício, de função ventricular direita e hemodinâmicos são combinados para definir o estado de baixo, intermédio ou alto risco, dependendo da mortalidade esperada de 1 ano. O actual algoritmo de tratamento fornece a estratégia inicial mais apropriada, incluindo monoterapia, terapia de dupla ou tripla combinação. É necessária uma maior escalada do tratamento se o estatuto de baixo risco não for alcançado nas visitas de acompanhamento programadas. Na maioria dos casos mais avançados, o transplante pulmonar pode ser necessário com a máxima terapia médica (Fig. 2).
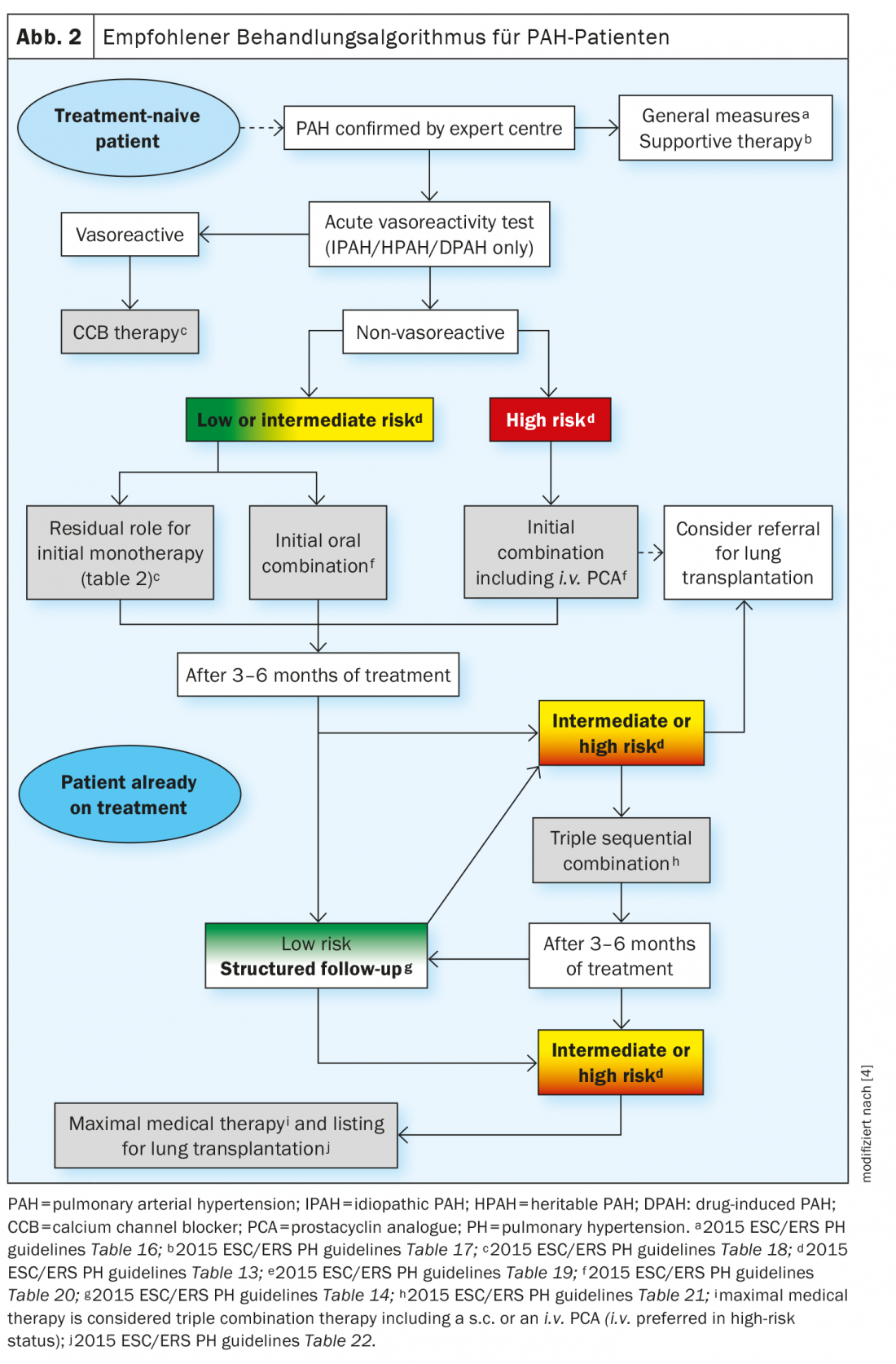
Os medicamentos de terapia oral aprovados incluem inibidores da fosfodiesterase 5 (PDE5i) tais como sildenafil e tadalafil, antagonistas dos receptores da endotelina (ERA) tais como bosentan, armbrisentan e macitentan, estimuladores da guanilato ciclase solúvel (sGC) tais como agonistas dos receptores do riociguat e da prostaciclina tais como selexipag. Também é possível tratar a HAP com terapia inalatória ou infusões contínuas.
Estudos prospectivos sobre terapia
Por exemplo, os inibidores de riociguat e fosfodiesterase-5 (PDE5i), que são aprovados para o tratamento da hipertensão arterial pulmonar (HAP), actuam através de diferentes mecanismos na mesma via. O ensaio REPLACE investigou portanto se o riociguat poderia ser uma opção alternativa para doentes com HAP que não respondem adequadamente ao tratamento com PDE5i. O objectivo era avaliar os efeitos da mudança da terapia PDE5i para o riociguat em comparação com a terapia PDE5i continuada em doentes com HAP com risco intermédio de mortalidade de 1 ano. Os resultados mostram que a passagem do tratamento com PDE5i para o riociguat, ambos actuando através da via de sinalização entre a guanilato ciclase solúvel em óxido nítrico e a guanosina monofosfato cíclica, pode ser uma opção estratégica para a escalada do tratamento em doentes com HAP com risco intermédio de mortalidade de 1 ano [5].
Sotatercept, um novo antagonista dos activistas, liga activinas e factores de diferenciação do crescimento numa tentativa de restabelecer o equilíbrio entre as vias de sinalização promotoras de crescimento e inibidoras do crescimento. No ensaio PULSAR, 24 semanas de tratamento com sotatercept em doentes com hipertensão arterial pulmonar que estavam a receber terapia de fundo para a mesma resultaram numa redução da resistência vascular pulmonar [6]. O desempenho físico (medido por 6 minutos a pé) e os níveis NT-proBNP também melhoraram sob sotatercept, de acordo com o Prof. Olschewski.
“Sensation” dos EUA
O Prof. Olschewski teve uma “sensação” a relatar dos EUA: Nunca antes tinha havido uma terapia aprovada para o PH em doenças pulmonares (grupo 3). Isso agora mudou – pelo menos nos EUA. O treprostinil inalado foi aí utilizado pela primeira vez neste grupo de pacientes. O ensaio INCREASE controlado por placebo durante 16 semanas registou doentes com doença pulmonar intersticial e hipertensão pulmonar que foram administrados treprostinil inalado usando um nebulizador de ultra-sons com administração pulsada em até 12 respirações (72 μg total) quatro vezes por dia. Em comparação com placebo, o treprostinil inalado melhorou significativamente o desempenho físico dos pacientes, medido pelo teste de caminhada de 6 minutos [7]. Nos EUA, o ingrediente activo já foi aprovado pela FDA, mas se o fabricante também se candidatará à Agência Europeia de Medicamentos (EMA) num futuro próximo ou esperar por mais estudos está ainda em aberto, como explicou o perito. “Em qualquer caso, é um lampejo de esperança”.

Formação em movimento normalizada estabelecida com sucesso
No ensaio aleatório controlado EU-TRAIN de treino de exercício em pacientes com hipertensão arterial pulmonar (HAP) e hipertensão pulmonar tromboembólica crónica (HPTC), realizado em 11 centros em 10 países europeus numa grande população de pacientes, observou-se uma melhoria significativa e clinicamente significativa no parâmetro primário 6MGT e nos parâmetros secundários OMS-FC, QoL e consumo máximo de oxigénio [8]. O estudo mostrou pela primeira vez que um programa de exercício seguro e eficaz como complemento da terapia medicamentosa pode ser padronizado e implementado em diferentes países com diferentes sistemas de cuidados de saúde.
Fonte: Pneumo Update 2021: Hipertensão Pulmonar, 12.11.2021
Literatura:
- Hoeper, et al: Fenótipos idiopáticos de hipertensão arterial pulmonar determinados por análise de clusters a partir do registo do COMPERA. J Heart Lung Transplant 2020, doi: 10.1016/j.healun.2020.09.011.
- Rosenkranz, et al: Hipertensão pulmonar em HFpEF e HFrEF: Fisiopatologia, diagnóstico, abordagens de tratamento. Coração 2019, doi: 10.1007/s00059-019-4831-6.
- Zeder, et al: A resistência vascular pulmonar elevada prevê a mortalidade em pacientes com DPOC. Eur Respir J 2021, doi: 10.1183/13993003.00944-2021.
- Galiè, et al: Estratificação de risco e terapia médica da hipertensão arterial pulmonar. European Respiratory Journal 2019, doi: 10.1183/13993003.01889-2018.
- Hoeper, et al: mudança para a terapia riociguat versus terapia de manutenção com inibidores de fosfodiesterase-5 em doentes com hipertensão arterial pulmonar (REPLACE): um ensaio controlado multicêntrico, aberto, randomizado. Lancet Respir Med 2021, doi: 10.1016/S2213-2600(20)30532-4.
- Humbert, et al: Sotatercept for the Treatment of Pulmonary Arterial Hypertension. N Engl J Med 2021, doi: 10.1056/NEJMoa2024277.
- Waxman, et al: Treprostinil inalado em Hipertensão Pulmonar devido a Doença Pulmonar Intersticial. N Engl J Med 2021, doi: 10.1056/NEJMoa2008470.
- Grüning, et al: O treino de exercício padronizado é viável, seguro e eficaz na hipertensão pulmonar arterial pulmonar e tromboembólica crónica: resultados de um grande ensaio europeu multicêntrico randomizado e controlado. Eur Heart J 2021, doi: 10.1093/eurheartj/ehaa696.
InFo PNEUMOLOGIA & ALEROLOGIA 2022; 4(1): 20-22