As neuropatias imunes variam na apresentação clínica, curso e patogénese. Nos últimos anos, foram detectados auto-anticorpos contra as proteínas nodais e paranodais do anel de Ranvier lacing. Os auto-anticorpos podem causar danos axonais e levar à desmielinização.
As neuropatias imunes variam na apresentação clínica, curso e patogénese e representam aproximadamente 9% de todas as polineuropatias [1]. Nos últimos anos, foram detectados auto-anticorpos contra as proteínas nodais e paranodais do anel de Ranvier lacing. Autoanticorpos contra neurofascina (NF), contactina 1 (CNTN1) ou proteína associada à contactina 1 (CASPR1) poderiam ser detectados em 10% dos doentes com polineuropatia inflamatória desmielinizante crónica (CIDP) [2]. Os auto-anticorpos podem causar danos axonais e levar à desmielinização [4]. Os pacientes seropositivos com CIDP diferem dos pacientes seronegativos no fenótipo clínico, nos resultados electrofisiológicos e na resposta à terapia padrão [2].
A detecção de auto-anticorpos nodais e paranodais específicos pode ajudar a encontrar terapias eficazes. Além do CIDP, os auto-anticorpos associados à doença também são conhecidos noutras neuropatias imuno-mediadas, como a síndrome de Guillain-Barré (GBS) com os subtipos polineuropatia inflamatória desmielinizante aguda (AIDP), neuropatia motora axonal aguda (AMAN) e neuropatia motora e sensorial aguda axonal (AMSAN), Síndrome de Miller-Fisher (MFS), neuropatia motora multifocal (MMN), neuropatia anti-MAG MGUS (neuropatia paraproteémica), neuronopatia sensível em doenças imunitárias sistémicas e polineuropatias paraneoplásicas.
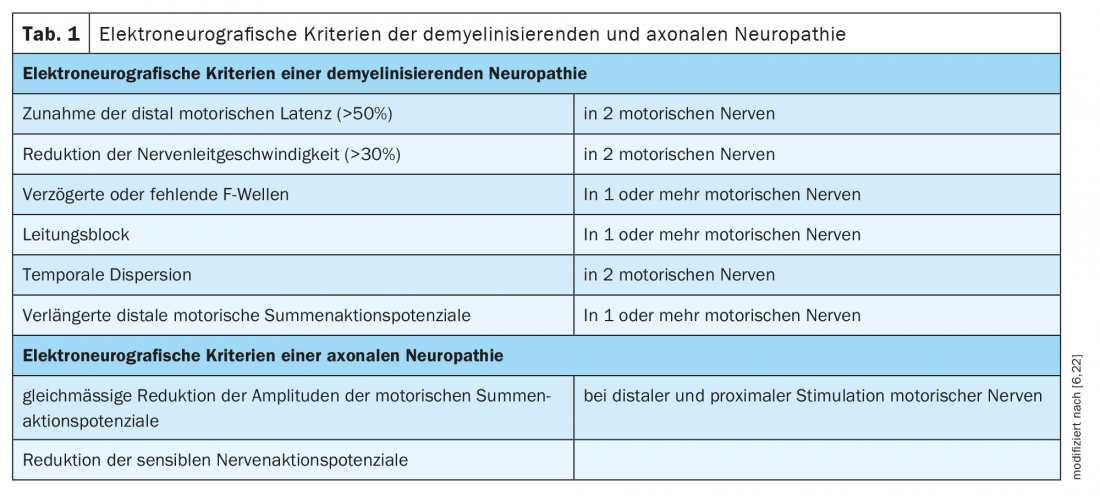
Patofisiologia e alterações electrofisiológicas de nodo- e paranodopatias
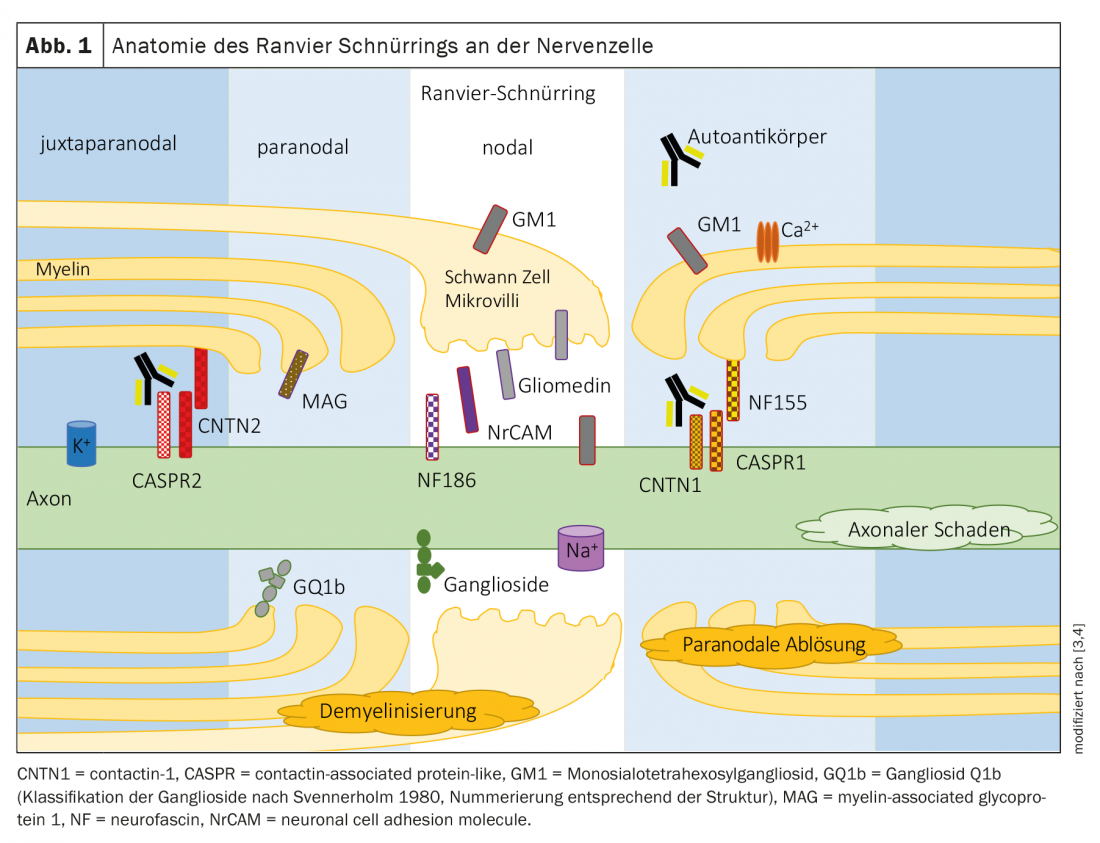
As polineuropatias são tradicionalmente divididas em desmielinizantes e neuropatias axonais de acordo com critérios em electrofisiologia (Tab. 1). Espera-se que a patologia subjacente esteja na célula de mielina/swan ou axônio em conformidade. As proteínas da região nodal ou paranodal do anel de laço de Ranvier são junções entre a mielina/microvelha da célula Schwan e o axônio (Fig. 1) . Em neuropatias devidas a auto-anticorpos contra proteínas nodais e paranodais, não é possível uma atribuição clara a um padrão desmielinizante ou axonal devido ao patomecanismo subjacente. O termo nodo- e paranodopatias foi proposto por Uncini, Susuki e Yuki em 2013 [8,9].
As características de nodo- e paranodopatia são
- Os anticorpos contra as proteínas nodais e paranodais têm diferentes etiologias, mas todos levam a disfunções na região nodal do anel de Ranvier.
- Continuum patológico desde o bloco de condução transitória até à degeneração axonal.
- Bloco de condução devido ao descolamento da mielina do axônio, alargamento da região nodal ou disfunção/disrupção dos canais de sódio (alta densidade na região nodal para condução salina) com polarização anormal segmentar do axolem.
- O bloco de condução pode ser rapidamente reversível sem o aparecimento de uma dispersão temporal significativa, o bloco de condução também pode persistir.
- A degeneração axonal depende da doença específica e da gravidade da doença, possivelmente seguida de um bloqueio de condução.
- O diagnóstico pode ser feito através de repetidos exames electrofisiológicos:
- a. na doença aguda, no caso de um bloqueio de condução rapidamente reversível ou redução da velocidade da condução nervosa sem dispersão temporal marcada ou progressão de um bloqueio de condução para degeneração axonal;
- b. em doenças crónicas com bloqueio de condução persistente e sinais adicionais de degeneração axonal.
A utilização do termo nodo- e paranodopatias coloca o foco na localização do dano nervoso primário e ajuda a melhor classificar a neuropatia desmielinizante segmentar, o paradoxo do dano axonal reversível e o bom prognóstico, apesar das anomalias axonais em electrofisiologia [9].
Autoanticorpos em polineuropatia desmielinizante crónica inflamatória (CIDP)
Localização: Na região paranodal, o complexo de contactin 1 e CASPR1 no axônio e neurofascina 155 na célula glial da bainha da mielina consolida o contato entre os processos da mielina e o axônio (Fig. 1) [10].
Anti-Contactoin 1 (CNTN1) IgG
- Sintomas: 20 – 49% mostram um início subagudo da doença. É conhecido um início de sintomas agressivo do tipo GBS com paresia subaguda de início (casuística). Uma neuropatia predominantemente motora é típica e a ataxia sensorial é frequentemente observada. A ocorrência de tremor é mais frequente do que no CIDP seronegativo, mas menos frequente do que no CIPD anti NF155-positivo. A idade de início é de cerca de 50-60 anos. Ano de vida. Globalmente, foi descrita uma idade mais elevada no início da doença para doentes CIDP anti-CNTN1 positivos do que para doentes CIDP seronegativos [2].
- Descobertas: Os exames electoneurográficos revelam alterações axonais no início do curso da doença (Tab. 1) .
- Implicações do tratamento: Os doentes respondem inadequadamente às imunoglobulinas intravenosas (IVIg). Os corticosteróides têm um bom efeito. Se houver uma resposta insuficiente à terapia padrão, a terapia de esgotamento das células B com rituximab é eficaz [14]. Deve-se notar que é importante manter a duração da doença tão curta quanto possível para que os danos axonais sejam minimizados [8]. Sob imunoterapia eficaz, os títulos séricos de anti CNTN1 diminuem [11].
Proteína associada à anti-contato 1 (CASPR1) IgG
- Sintomas: A dor neuropática pronunciada foi descrita em doentes seropositivos anti CASPR1. Os sintomas geralmente começam subactualmente com uma expressão severa e são inicialmente motorizados. O Anti CASPR1 foi detectado em doentes com CIDP e GBS. O início da doença é dado num relatório de caso por volta dos 30 anos de idade.
- Descobertas: Os blocos de condução reversível podem ser detectados nas investigações electrofisiológicas [11].
- Implicações do tratamento: A falta de resposta ao IVIg e à metilprednisolona tem sido descrita. O Rituximab tem demonstrado uma eficácia muito boa. A dor neuropática associada melhora com uma terapia eficaz [2].
Anti Neurofascin 155 (NF155) IgG
- Sintomas: Tipicamente, uma elevada proporção de pacientes CIDP NF155-positivos tem acentuado distintamente a fraqueza. Outros sintomas podem incluir tremor de alta amplitude e baixa frequência e ataxia sensorial com sinais cerebelares. A associação com HLA-DRB1*15 foi descrita [12]. A idade de início é significativamente mais jovem do que no CIPD seronegativo, com uma idade de início de cerca de 20-30 anos.
- Descobertas: O exame electrofisiológico mostra um padrão desmielinizante com um claro prolongamento das latências distais e das latências das ondas F (Tab. 1). Foi detectado um forte aumento de proteínas no líquido cefalorraquidiano [13].
- Implicações do tratamento: A resposta ao IVIg é fraca. Foi descrita uma melhoria parcial nos corticosteróides. Um bom sucesso terapêutico pode ser esperado com rituximab e plasmapheresis [14]. Os títulos séricos de anti NF155 diminuem sob imunoterapia eficaz, e foi apresentada uma correlação com a melhoria clínica [11].
Anti Neurofascina 140/186 (NF 140/186) IgG
- Sintomas: Pacientes presentes com – uma poliradi-culopatia sensorimotora simétrica com um curso severo. Pode também ocorrer ataxia sensorial e envolvimento do nervo craniano. Alguns doentes apresentavam concomitantemente doença auto-imune. A idade de início é de cerca de 50-60 anos. descrito durante o primeiro ano de vida [2].
- Descobertas: A electroneurografia revelou descobertas desmielinizantes com blocos de condução (em 3/5 pacientes) e características axonais (em 2/5 pacientes) [15].
- Implicações do tratamento: No IVIg e nos corticosteróides, os sintomas melhoraram parcialmente. Uma resposta potencialmente boa tem sido descrita ao rituximab [14].
Autoanticorpos em polineuropatias inflamatórias agudas – Síndrome de Guillain-Barré (GBS) com os subtipos AIDP, AMAN e AMSAN
Localização: Os canais iónicos do anel de ligação de Ranvier são estabilizados por gangliosídeos, glicosfingolípidos contidos na membrana celular das regiões nodais e paranodais [16]. Os diferentes subtipos da síndrome de Guillain-Barré estão cada um associado com autoanticorpos a diferentes gangliosides e diferem em descobertas electrofisiológicas.
A poli-neuropatia desmielinizante inflamatória aguda (AIDP) é o subtipo mais comum de GBS na Europa e América do Norte. Normalmente, não podem ser detectados aqui autoanticorpos específicos.
- Sintomas: A doença é frequentemente precedida por uma infecção gastrointestinal ou respiratória. Inicialmente, a hispestesia, paraestesia, paresia e dor ocorrem nas extremidades. A paresia é bilateral, simétrica e progressiva. Os sintomas motores desenvolvem-se dentro de 12 horas a 28 dias até à tetraparese com envolvimento dos músculos respiratórios. Além disso, podem ocorrer défices nervosos cranianos e disfunções autonómicas [4].
- Constatações: Este subtipo desmielinizante de polineuropatia inflamatória aguda caracteriza-se por uma marcada redução da velocidade de condução nervosa, aumento das latências distais do motor, bloqueios de condução, dispersão temporal anormal, latências prolongadas das ondas F ou ausência de ondas F [17].
Anti LM1 (sialosylneolactotetraosylceramide)
Autoanticorpos contra LM1 (sialosylneolactotetraosylceramide) causam polineuropatia desmielinizante inflamatória aguda (AIDP) se forem monoespecíficos. Foram descritos casos em que anticorpos anti LM1 IgG provocam uma reacção cruzada com gangliosides tais como GM1, GalNAc-GD1a, GD1b e GQ1b, para que neste caso se possa fazer o diagnóstico de AMAN ou AMSAN.
Anti GalC (Galactocerebroside)
Autoanticorpos contra galactocerebrosides (GalC) foram detectados em crianças com polineuropatia inflamatória aguda desmielinizante grave associada à M. pneumonia (AIDP) [18].
Anti Neurofascin 186, Anti Gliomedin, Anti NrCAM, Anti CNTN1, Anti Neurofascin 155, Anti CASPR1 e Anti CASPR2
Em estudos, os autoanticorpos nodais anti neurofascina 186, anti gliomedina e anti NrCAM, e os autoanticorpos paranodais anti CNTN1, anti neurofascina 155 e anti CASPR1 (ver acima na secção CIDP) foram detectados em doentes adultos com GBS. Os auto-anticorpos justaparanodais ao CASPR2 foram descritos em 2 casos de GBS em crianças.
A neuropatia axonal motora aguda (AMAN) ocorre principalmente na Ásia [19].
- Sintomas: Correspondente à AIDP (ver acima) sem perturbações sensoriais.
- Constatações: Este subtipo axonal de polineuropatia inflamatória aguda caracteriza-se por uma redução acentuada das amplitudes do potencial de acção cumulativa motora (CMAP) e das chamadas “falhas reversíveis de condução”, ou seja, a redução da amplitude CMAP e dos blocos de condução podem recuperar subitamente em repetidos exames electroeurográficos sem evidência de dispersão temporal como sinal de remineelinização. [16].
Anti-GM1 IgG, Anti-GM2, Anti-GD1b IgG, Anti-GT1b, Anti- GM3, Anti-GD1a IgG e Anti-GalNac-GD1a
Os anticorpos ganglioside anti-GM1 IgG, anti-GM2, anti-GD1b, anti-GT1b, anti-GM3, anti-GD1a IgG e anti-GalNac-GD1a podem ser detectados em cerca de 80% dos doentes com AMAN. Estes auto-anticorpos podem ocorrer isoladamente ou em combinação.
Neuropatia aguda motora e sensorial axonal (AMSAN)
- Sintomas: ver AIDP
- Conclusões: Os mesmos critérios que AMAN mais redução da amplitude do potencial de acção do nervo sensorial (Unicini et al. 2018).
Anti-GM1 IgG, anti-GM1b, anti-GD1a IgG
Os anticorpos gangliosídeos anti-GM1, anti-GM1b, anti-GD1a são detectáveis na neuropatia motora e sensorial axonal aguda (ASMAN) [19].
Autoanticorpos na Síndrome de Miller Fisher (MFS)
Localização: Ganglioside GQ1b encontra-se principalmente na mielina paranodal dos nervos cranianos que fornecem os músculos -eye [19].
Anti GQ1b IgG
- Sintomas: Oftalmoplegia aguda, neuropatia atáxica aguda e areflexia são os principais sintomas da síndrome de Miller Fisher. Na encefalite do tronco cerebral de Bickerstaff, há oftalmoplegia e ataxia, consciência diminuída e geralmente hiperreflexia.
Os auto-anticorpos GQ1b podem ser detectados em 90% dos doentes com síndrome de Miller Fisher (MFS). A síndrome de Miller-Fisher e a encefalite do tronco cerebral de Bickerstaff estão associadas ao anti GQ1b, pelo que Shahrizaila e Yuki agrupam as doenças como síndrome do anticorpo anti-GQ1b [20].
Autoanticorpos em neuropatia motora multifocal (MMN)
Localização: Ganglioside GM1 é principalmente localizada na região nodal do anel de laço de Ranvier. Os auto-anticorpos ligam-se na região nodal e activam o complemento, afectando a agregação dos canais de sódio.
Complexo Anti-Galactocerebroside Anti GM1 IgM e Anti GM1-Galactocerebroside
- Sintomas: A paresia das extremidades aumenta lentamente, na sua maioria assimétrica é característica, muitas vezes começando nas extremidades superiores. Não são encontrados défices sensíveis. Além disso, podem ocorrer tremores, fasciculações e cãibras.
- Descobertas: anticorpos anti-GM1 IgM são detectáveis em 50% de todos os doentes com MMN [21]. Os blocos de condução podem ser detectados em exames electrofisiológicos. A activação do complemento pode causar danos axonais.
- Implicações do tratamento: Boa resposta ao IVIg. A detecção de auto-anticorpos pode apoiar o diagnóstico de MMN se nem todos os critérios forem cumpridos e ajudar a iniciar uma terapia eficaz com o IVIg. Clinicamente, existe uma doença do 2º neurónio motor, tornando a MMN um importante diagnóstico diferencial de esclerose lateral amiotrófica [11].
Autoanticorpos em polineuropatia de MGUS (MGUS-P)
Localização: A glucoproteína associada à mielina (MAG) é localizada na mielina da região paranodal.
Anti MAG (glucoproteína associada à mielina) IgM
- Sintomas: É característica uma polineuropatia distal atáxica, predominantemente sensível, lentamente progressiva. As extremidades distais superiores são frequentemente afectadas [11]. A maioria dos doentes tem menos de 50 anos de idade.
- Descobertas: Um padrão desmielinizante é detectável no exame electrofisiológico. A imunofixação revela a gamopatia monoclonal IgM. Os anticorpos anti-MAG IgM são positivos em 50% dos doentes com MGUS-P. O nível do título de anticorpos não parece estar correlacionado com a gravidade da doença e a resposta à terapia. A detecção de autoanticorpos anti-MAG é apenas necessária para o diagnóstico.
- Implicações do tratamento: Alguns estudos mostraram uma resposta à plasmaferese, ciclofosfamida, IVIg e rituximab. Há provas de que o esgotamento precoce das células B com rituximab pode influenciar a progressão [11].
Autoanticorpos associados a doenças imunitárias sistémicas
anticorpos SSA (Ro) e SSB (La), anticorpos antineuronais, anticorpo FGFR3 (receptor de factor de crescimento fibroblasto 3)
- Sintomas: Em pacientes (mais jovens) com um curso progressivo, agudo ou subagudo de neuropatia/neuronopatia sensorial, é útil uma pesquisa da síndrome de Sjögren ou um rastreio de doenças auto-imunes [22].
- Descobertas: No QCA há um ligeiro aumento de proteínas com contagem de células normal. Electrofisiologicamente, as anomalias nos nervos sensíveis que não são dependentes do comprimento são detectáveis. A perda extensiva ou baixa amplitude dos potenciais de acção nervosa sensorial é típica, tal como uma distribuição clínica assimétrica (“irregular”). Ocasionalmente, os nervos sensíveis dos braços podem ser mais afectados do que os das pernas [23].
- Implicações terapêuticas: A doença subjacente deve ser tratada.

Em que apresentação clínica se deve pensar numa génese mediada por autoanticorpos?
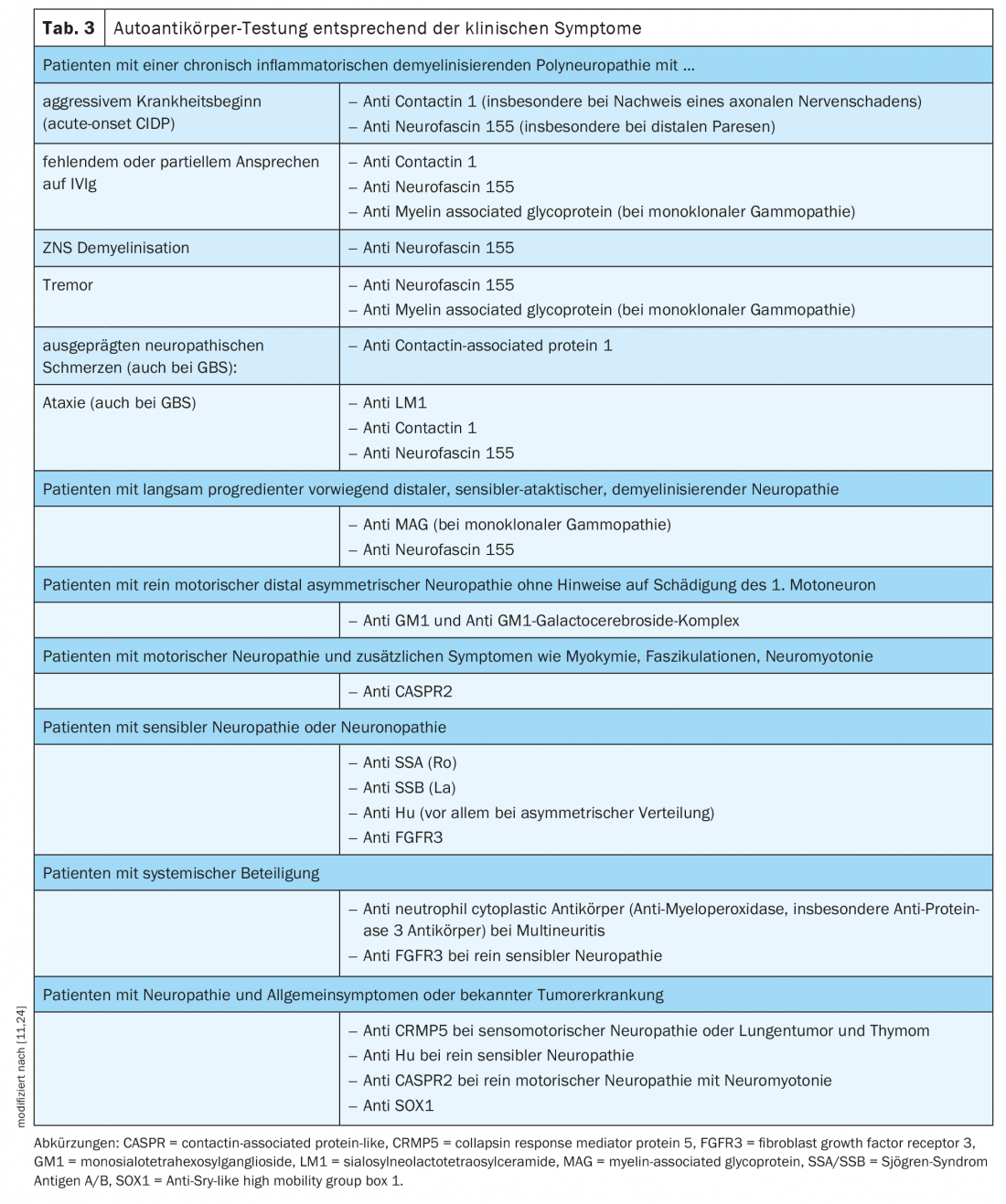
Os testes para auto-anticorpos nodais e paranodais devem ser realizados em doentes com um curso agudo, subagudo ou crónico de polineuropatia desmielinizante adquirida associada a sintomas adicionais tais como tremor, envolvimento distal ou má resposta à IVIg [11] (Tabela 3).
Porque é que a terapia padrão é frequentemente inadequada para a polineuropatia desmielinizante inflamatória crónica seropositiva?
Estudos demonstraram que os auto-anticorpos nodais e paranodais pertencem a diferentes subclasses de IgG (Tabela 4) . IgG2 e IgG3 foram encontrados em cursos monofásicos anti CNTN1 e CASPR1 seropositivos GBS, IgG4 apenas em cursos crónicos. De Appeltshauser et al. foi descrito que no decurso da doença pode ocorrer uma mudança de classe de IgG3 para IgG4 na neuropatia seropositiva anti CNTN1 e anti CASPR1 [25]. Actualmente, as subclasses IgG de auto-anticorpos nodais e paranodais só são determinadas em laboratórios de investigação.

As neuropatias mediadas por IgG3 respondem bem à terapia padrão IVIg. O IVIg é eficaz em polineuropatias inflamatórias, particularmente através da inibição do complemento e neutralização de anticorpos. Os doentes com anticorpos IgG2 e IgG4 ou não mostram melhorias com infusões de IVIg ou a resposta diminui ao longo do curso da doença. A resposta fraca ou ausente ao IVIg nas doenças mediadas por IgG4 é atribuída à baixa capacidade de ligar receptores FcγIIb e à falta de activação do complemento [14]. Nas doenças neurológicas e não neurológicas auto-imunes mediadas por IgG4, tais como a miastenia gravis positiva antimúsculo, a pancreatite auto-imune e a colangite esclerosante, a eficácia do rituximab tem sido comprovada em numerosos estudos. O efeito de esgotamento das células B do rituximab é crucial na doença mediada por IgG4. Foi relatada uma resposta parcial de doentes com anticorpos IgG4 nodal e paranodal a corticosteróides [14]. Nas neuropatias autoanticorpositivas, é necessária uma terapia rápida e eficaz antes que ocorram danos irreversíveis. Na ausência de resposta à terapia padrão (IVIg & esteróides) para a polineuropatia inflamatória crónica desmielinizante, a terapia com rituximab deve ser considerada.
Mensagens Take-Home
- Dentro das neuropatias imuno-mediadas, os nodo-/paranodopatias devem ser considerados para diagnóstico diferencial, uma vez que aqui existem opções terapêuticas eficazes.
- A química laboratorial pode detectar autoanticorpos contra proteínas nodais e paranodais do anel de Ranvier em nodo- e paranodopatias.
- As características electroneurográficas são definidas para nodo- e paranodopatias.
- A base do diagnóstico de neuropatias associadas ao auto-anticorpo são os exames eletroneurográficos (desmielinização ou padrão axonal, características de nodo- e paranodopatias) e o diagnóstico laboratorial e do LCR (exclusão de diagnósticos diferenciais relevantes, detecção da dissociação da cialbumina no LCR); a sonografia do nervo (padrão de inchaço do nervo) também é útil.
- Testar auto-anticorpos nodais e paranodais de soro (sem produção intratecal e baixos títulos no CSF).
- Na ausência de melhoria com terapia de primeira linha (IVIg, esteróides) e em pacientes com um curso agudo ou subagudo de polineuropatia desmielinizante adquirida com sintomas adicionais tais como tremor, ataxia e envolvimento distal (ver Quadro 2: Bandeiras vermelhas no CIDP), considerar a determinação de auto-anticorpos e, se necessário, a terapia com rituximab.
- Selecção de auto-anticorpos com base em sintomas clínicos (Tab. 3).
Literatura:
- Visser NA: Incidência de polineuropatia em Utrecht, Países Baixos. Neurologia 2015, 20 de Janeiro; 84(3): 259-264.
- Vural A, Doppler K: Autoanticorpos contra o nó de Ranvier em polineuropatia inflamatória crónica desmielinizante seropositiva: relevância diagnóstica, patogénica, e terapêutica. Frente. Imunol. 2018, 14 de Maio; 9: 1029
- Stathopoulos P: alvos antigénicos auto-imunes no nó de Ranvier em perturbações desmielinizantes. Nat Rev Neurol. 2015 Mar; 11(3): 143-156.
- Kieseier BC, Mathey EK, Sommer C: Neuropatias imunizadas. Nat Rev Dis Primers 2018 4, 31.
- Svennerholm L: designação Ganglioside. Adv Exp Med Biol. 1980; 125: 11
- Grether NB, et al: Diagnóstico de polineuropatias imuno-mediadas. DGNeurologia 2020; 3 (2): 147-158.
- Uncini A, Kuwabara S: Critérios de electrodiagnóstico para a síndrome de Guillain-Barre: uma revisão crítica e a necessidade de uma actualização. Clin Neurophysiol, 2012, 123(8), 1487-1495.
- Uncini A, Kuwabara S: Nodopatias do nervo periférico: um conceito emergente. J Neurol Neurosurg Psychiatry, 2015, 86(11), 1186-1195.
- Uncini A, Susuki K, Yuki N: Nodo-paranodopatia: para além da classificação desmielinizante e axonal em neuropatias mediadas por anticorpos anti-ganglioside. Clin Neurophysiol, 2013, 124(10), 1928-1934.
- Poliak S, Peles E: A diferenciação local dos axônios mielinizados nos nós de Ranvier. Nat Rev Neurosci, 2003, 4(12), 968-980.
- Querol L: Autoanticorpos em neuropatias inflamatórias crónicas: implicações diagnósticas e terapêuticas, Nat Rev Neurol. Neurologia, 2017, Set; 13(9): 533-547.
- Martinez-Martinez L: Anti-NF155 poliradiculoneuropatia inflamatória crónica desmielinizante fortemente associada à HLA-DRB15. J Neuroinflamação 2017; 14: 224.
- Kadoya M: IgG4 anti-neurofascina155 anticorpos em poliradiculoneuropatia desmielinizante crónica inflamatória: Significado clínico e utilidade diagnóstica de um ensaio convencional. Journal of Neuroimmunology 2016 Dez 15; 301: 16-22.
- Bunschoten C: Progress in diagnosis and treatment of chronic inflammatory demyelinating polyradiculoneuropathy, Lancet Neurology 2019; 18: 784-94.
- Delmont E, Manso C, Querol L, et al: Autoanticorpos a isoformas nodais de neurofascina em polineuropatia inflamatória desmielinizante crónica. Cérebro. 2017 Jul 1;140(7): 1851-1858.
- Susuki K: Os gangliosides contribuem para a estabilidade das junções paranodais e dos aglomerados de canais iónicos em fibras nervosas mielinizadas. Glia, 2007a, 55(7): 746-757.
- Uncini A, Kuwabara S: O electrodiagnóstico dos subtipos da síndrome de Guillain-Barré: Qual é a nossa posição? Neurofisiologia clínica. 2018 Dez; 129(12): 2586-2593.
- Meyer Sauteur PM: Mycoplasma pneumoniae desencadeando a síndrome de Guillain-Barré: Um estudo de controlo de casos, Ann Neurol 2016 Out; 80(4): 566-580.
- Pei S: Variantes axonais da síndrome de Guillain-Barré: uma actualização. Springer Nature 2020 Março.
- Shahrizaila N, Yuki N: encefalite do tronco cerebral de Bickerstaff e síndrome de Fisher: síndrome dos anticorpos anti-GQ1b. J Neurol Neurosurg Psychiatry 2013, 84: 576-583.
- Van Asseldonk JT: Neuropatia motora multifocal. Lancet Neurol, 2005, 4(5): 309-319.
- Heuss D: Diagnostik bei Polyneuropathien, S1-Leitlinie, 2019, in: Deutsche Gesellschaft für Neurologie (Hrsg.), Leitlinien für Diagnostik und Therapie in der Neurologie. Em linha: www.dgn.org/leitlinien. (recuperada a 01.04.2021).
- Sghirlanzoni A: Doenças sensoriais dos neurónios. A Neurologia Lancet. 2005; 4(6): 349-361
- Sol X: Anticorpos Anti-SOX1 na Síndrome Neurológica Paraneoplásica. J Clin Neurol. 2020 Oct;16(4):530-546.
- Appeltshauser L: Anticorpos antiparanodais e subclasses de IgG em neuropatia auto-imune aguda. Neurol Neuroimmunol Neuroinflamm. 2020 Jul 24; 7(5): e 817.
- Ilyas A: Distribuição de imunoglobulina G subclasse de auto-anticorpos a gangliosídeos em doentes com síndrome de Giullain-Barre. Res Commun Mol Pathol Pharmacol. 2001 Jul; 109(1-2): 115-123.
- Lardone RD: Desordens neurológicas – associados a anticorpos IgG antiglicosfingolípidos apresentam uma distribuição de subclasse IgG diferentemente restrita. Rep. Ciência 2020 Ago 4;10(1): 13074.
InFo NEUROLOGIA & PSYCHIATry 2021; 19(3): 19-25.