Existem muitas causas diferentes e por vezes raras de feridas crónicas. O diagnóstico correcto é crucial para um tratamento adequado. Uma biópsia é frequentemente útil para causas raras e pouco claras. O diagnóstico e terapia de feridas devem ser efectuados como parte de cuidados colaborativos multimodais.
Se as causas de pacientes com feridas não cicatrizantes não forem identificadas, isto pode ter consequências muito graves, explicou o Prof. Dr. med. Joachim Dissemond, Hospital Universitário de Essen (D), na conferência anual da Associação Austríaca de Feridas (AWA). “O diagnóstico e a terapia devem definitivamente ser realizados de forma interdisciplinar e interprofissional”, apela o orador e sublinha que a biopsia tem um significado muito importante para o diagnóstico de causas raras de feridas [1]. A fim de determinar factores desencadeantes ou envolvimento extracutâneo, um historial médico completo e testes laboratoriais seleccionados também fazem parte do procedimento de clarificação diagnóstica. As doenças órfãs são descritas na base de dados da Orphanet [2].
A fim de melhorar a qualidade de vida dos doentes com feridas crónicas e de combater as complicações de feridas não cicatrizantes, a gestão moderna de feridas e, se necessário, a terapia da dor são indicadas como medidas de acompanhamento.
Angite leucocitocítica cutânea – púrpura palpável como sintoma principal
A angite leucocitocítica cutânea é a forma mais comum de vasculite cutânea. A doença pode ocorrer em qualquer idade e manifesta-se tipicamente como púrpura palpável e/ou petéquias nas extremidades inferiores com distribuição unifocal ou multifocal. A vasculite está associada à inflamação neutrofílica predominantemente confinada às vênulas cutâneas pós-capilares superficiais, sem vasculite sistémica ou glomerulonefrite.
O eritema lívido ocorre frequentemente, o que é uma indicação de que a inflamação está presente, explica o Prof. Dissemond. Ocorrem necrose e geralmente múltiplas úlceras. Normalmente ambas as pernas são afectadas. “Puremente unilateral seria extremamente invulgar para a angite leucocitocítica cutânea”, disse o orador [1].
O diagnóstico de angite leucocitocítica cutânea é um diagnóstico de exclusão. Em geral, as amostras de biopsia de pele de lesões com idades entre 24 e 48 horas devem ser examinadas com microscopia ligeira e imunofluorescência directa. As causas comuns desta doença rara são infecções e/ou drogas, sendo até 50% dos casos idiopáticos. Como opção de tratamento clássica, o Prof. Dissemond menciona corticosteróides sistémicos, por exemplo prednisolona 1 mg por kg/KG.
Pyoderma gangraenosum – Paracelsus facilita o diagnóstico
O pioderma gangraenosum caracteriza-se por úlceras recorrentes da pele com exsudado muco-purulento ou hemorrágico. Começa frequentemente com uma pústula esterilizada. O pus é estéril porque é granulócito neutrofílico, explica o Prof. Dissemond [1]. A incidência é maior na faixa etária dos 20-50 anos, sendo as mulheres mais frequentemente afectadas do que os homens. Típica desta dermatose neutrofílica são as margens minadas e o eritema sombrio-lívido. Em cerca de 70% dos doentes, os lados extensores das extremidades inferiores (canela) são afectados, mas também são possíveis outras localizações. As comorbidades comuns são doenças inflamatórias crónicas do intestino (doença de Crohn, colite ulcerativa), mas as neoplasias também não são incomuns nestes pacientes. “É uma doença potencialmente paraneoplásica”, disse o orador [1]. Portanto, os doentes com pioderma gangraenosum, especialmente se tiverem um curso de terapia-refractária, devem ser examinados especialmente para neoplasias hematológicas (por exemplo, síndrome mielodisplásica). Como se diagnostica o pioderma gangraenosum? A classificação como diagnóstico de exclusão já não está actualizada, sublinha o Prof. Dissemond e acrescenta: “Hoje, é possível fazer o diagnóstico com relativamente poucas medidas” [1]. É útil utilizar a pontuação Paracelsus recomendada nas directrizes alemãs (Tab. 1). O orador refere a imunossupressão como as medidas terapêuticas mais importantes. O tratamento mais bem documentado é o tratamento sistémico com corticosteróides e ciclosporina A.

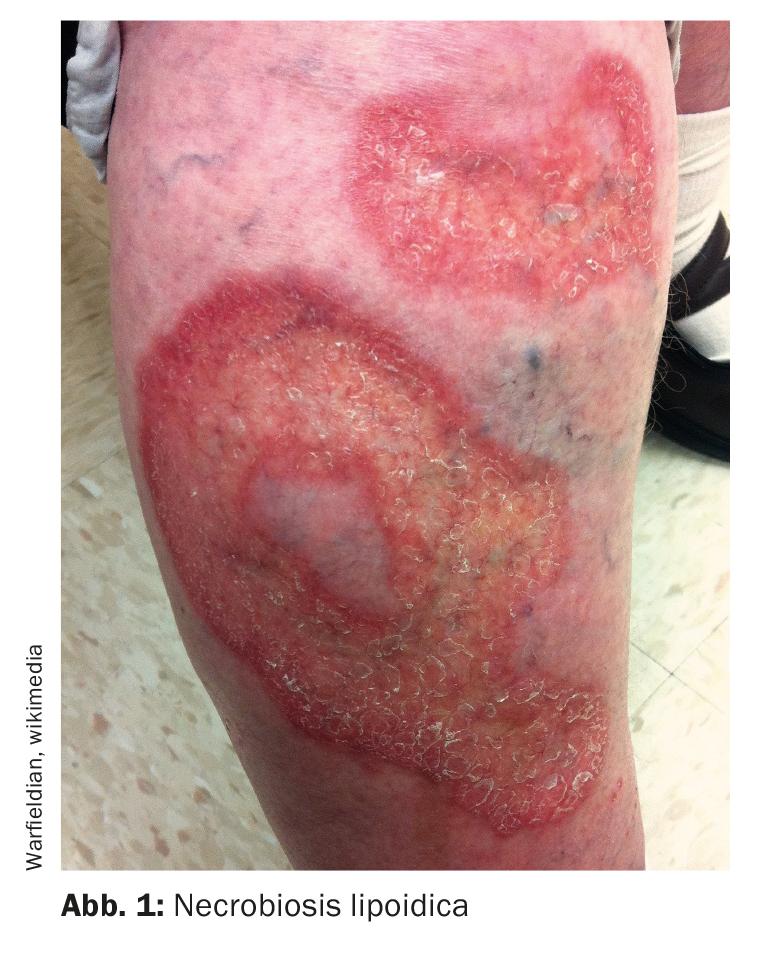
Necrobiosis lipoidica – as ulcerações são extremamente dolorosas
A necrobiose lipoidica (Fig. 1) é uma doença granulomatosa rara, cuja etiologia ainda é mal compreendida. Os lados extensores das extremidades inferiores são também o local de predilecção para esta doença órfã – em até 90% dos casos, as manifestações da doença são localizadas nesta área. Um sinal clínico típico é uma placa amarelo-laranja com atrofia e telangiectasia. “Não ulcera necessariamente, mas quando ulcera, dói terrivelmente”, diz o Prof. Dissemond [1]. Contudo, se não houver ulcerações, a necrobiose lipoidica é quase imperceptível para os pacientes. Normalmente começa com uma perna, a segunda perna segue-se após alguns meses ou anos. Uma biópsia é muito importante para o diagnóstico. O nome anterior era necrobiosis lipoidica diabeticorum. Como sabemos hoje, no entanto, apenas 50% dos casos estão associados à diabetes mellitus, explicou o orador. Contudo, as pessoas afectadas têm um risco acrescido de desenvolver diabetes.
As opções de tratamento situam-se na área não rotulada. “Não há terapia aprovada”, disse o orador [1]. O tratamento com cortisona aplicada sistemicamente é delicado em diabéticos. Na literatura, encontram-se descrições do uso de ácido fumárico, e por vezes são também utilizados produtos biológicos (por exemplo, inibidores de TNF-alfa), acrescenta o Prof.
Livedovasculopatia – associada a parâmetros pró-coagulantes
A livedovasculopatia (Fig. 2)é uma doença vascular crónica recorrente. A trombose na microcirculação leva a uma perfusão reduzida e subsequente ulceração da pele. As ulcerações afectam exclusivamente a extremidade inferior, especialmente a região malleolar. A directriz S1 sobre o diagnóstico e tratamento da ovasculopatia vivida publicada em 2021 aponta que a confirmação histológica do diagnóstico só é possível na fase aguda da doença, a fase de isquemia [3]. Recomenda-se que seja retirado um exemplar suficientemente grande (de preferência uma biópsia do fuso) da área marginal da área afectada. As características são frequentemente difíceis de reconhecer os depósitos de fibrina nas paredes dos vasos, bem como os trombos de fibrina, que se encontram principalmente nos vasos da derme superior e média. Vários parâmetros procoagulatórios são descritos na vivovasculopatia. Estes incluem defeitos na activação do plasminogénio endotelial, disfunção plaquetária e aumento da formação de fibrina. A deposição de fibrina e a formação de trombos levam à isquemia dos tecidos e subsequentemente à ulceração. Atrofia pode desenvolver-se como uma manifestação crónica e ponto final de processos de remodelação de cicatrizes. Esta é uma cicatriz em forma de relâmpago ou estrela, semelhante a uma porcelana.
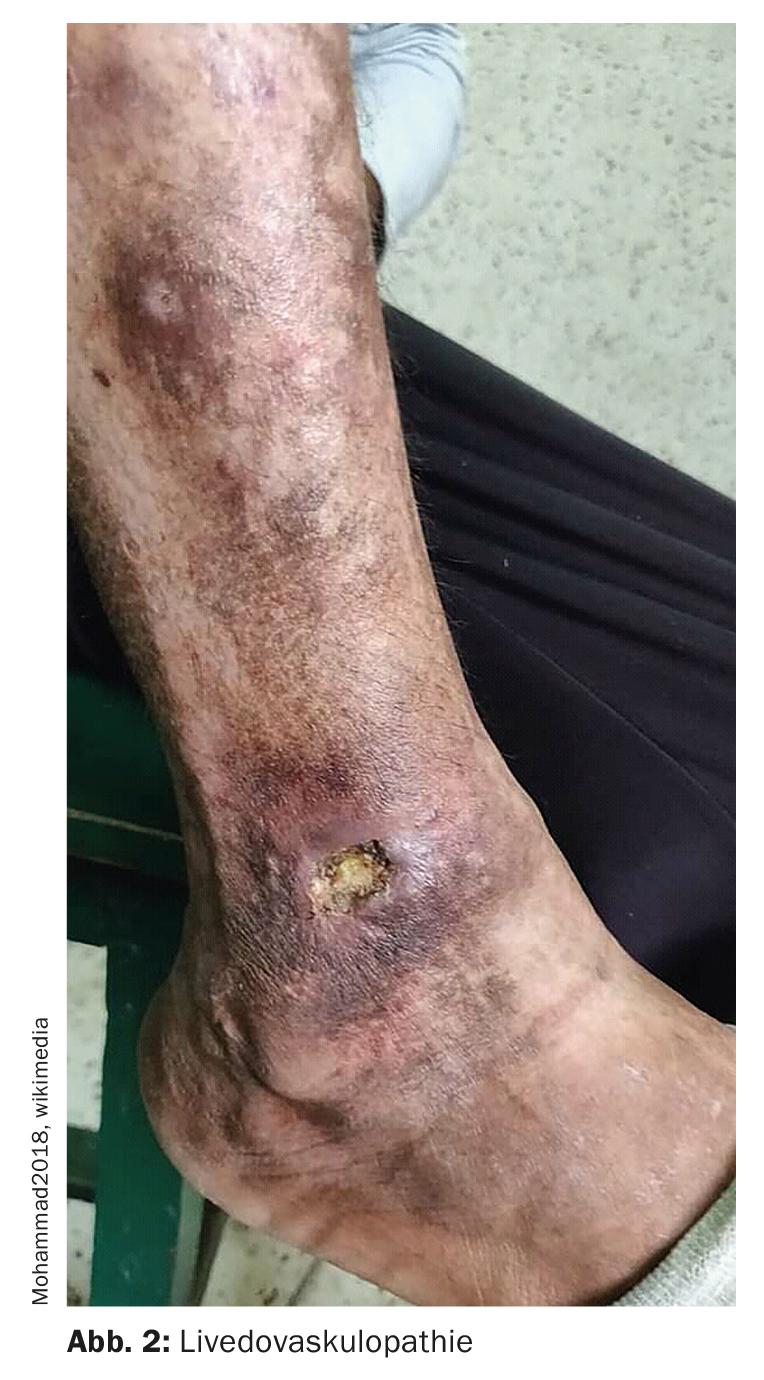
Para prevenir a progressão do enfarte crónico recorrente da pele com transformação cicatricial do local de manifestação, é necessário o início precoce da terapia medicamentosa. A estratégia de tratamento mudou em comparação com o passado. “Actualmente, os doentes são tratados reologicamente, o que significa heparina de baixo peso molecular”, explica o orador. Em poucos dias, os pacientes têm significativamente menos dor. Os DOAKs (anticoagulantes orais directos) são outra opção de tratamento. Na melhor das hipóteses, a cortisona pode ser adicionada nos primeiros dias, relata o Prof. Dissemond.
Calciflaxis – risco de vida se não tratada
Em 90-95% dos casos desta vasculopatia, também chamada “arteriolopatia uraémica calcificante”, envolve pacientes com insuficiência renal em fase terminal. O local de predilecção são as extremidades inferiores, as manifestações típicas são a necrose e o eritema lívido. A fase inicial caracteriza-se por uma endurecimento doloroso da pele, que pode assemelhar-se a uma neuralgia zoster [4]. A pele é frequentemente lívida avermelhada, com uma descoloração de padrão reticular. Típico é o desenvolvimento de um achado de palpação em couro, em forma de placa. O quadro completo é caracterizado por ulcerações profundas que excedem claramente a derme. A maior parte das vezes, o aro tem uma textura irregular, semelhante a um mapa. É uma condição médica grave. Terapêuticamente, uma tentativa de tratamento com tiossulfato de sódio pode ser útil. Em duas séries de casos não controlados com pacientes com calcificação (n=199), o tiossulfato de sódio foi utilizado como parte de uma abordagem terapêutica multimodal [5,6]. Foi administrada uma dose de 25 g de tiossulfato de sódio sob a forma de infusão no final / no final de cada hemodiálise. A duração do tratamento pode ser de semanas ou meses [4].
Síndrome de Klinefelter – aumento do risco de flebothrombose
Existem também causas genéticas de distúrbios graves de cicatrização de feridas. A análise genética é necessária para o diagnóstico. Na síndrome de Klinefelter, existe uma aberração cromossómica numérica sob a forma de trissomia (47, XXY). A incidência de flebothrombose é até 20 vezes maior do que na população normal devido a vários factores trombogénicos. As feridas crónicas correspondem geralmente a uma úlcera pós-trombótica da perna.
Congresso: Associação Austríaca de Feridas
Literatura:
- Dissemond J: Causas raras de feridas crónicas (“doenças órfãs”). Prof. Dr. med. Joachim Dissemond, Associação Austríaca de Feridas, 25.03.2022
- Orphanet, www.orpha.net (último acesso 17.08.2022)
- Görge T, et al: S1-Leitlinie Diagnostik und Therapie der Livedovaskulopathie, 2021; 013-098.
- Brandenburg VM, et al: Calciphylaxis. Dtsch Med Wochenschr 2015; 140: 347-351.
- Nigwekar SU, et al: Sodium thiosulfate therapy for calcific uremic uremic arteriolopathy. Clin J Am Soc Nephrol 2013; 8: 1162-1170.
- Zitt E, et al: Utilização de tiossulfato de sódio num ambiente multi -intervencional para o tratamento da calcificação em doentes em diálise. Nephrol Dial Transplant 2013; 28: 1232-1240.
- Hobbs MM, Ortega-Loayza AG: Pyoderma gangrenosum: Das perspectivas históricas às investigações emergentes. Int Wound J 2020; 17(5): 1255-1265.
PRÁTICA DA DERMATOLOGIA 2022; 32(4): 40-41
PRÁTICA DO GP 2022; 17(9): 16-17