As porfírias são um grupo de doenças metabólicas muito raras, geneticamente determinadas, com uma desordem de biossíntese da hemorragia. A tríade típica dos sintomas numa recaída consiste em dor abdominal, sintomas cerebrais e neuropatia periférica. Outro sinal importante é a descoloração vermelha da urina. Para o tratamento de um episódio agudo, a administração intravenosa de hemarginato (Normosang®) é a terapia de escolha. Ao cuidar dos doentes, é particularmente importante evitar os medicamentos que desencadeiam recaídas (cartão de emergência). Os doentes com porfíria recentemente diagnosticada devem ser incluídos no Registo de Porfírias Agudas de Munique.
As porfírias são um grupo de doenças metabólicas muito raras, geneticamente determinadas, que se baseiam numa desordem de biossíntese da hemorragia. No organismo humano, a hemoglobina é um componente da hemoglobina, responsável pelo transporte de oxigénio, e da mioglobina proteica armazenadora de oxigénio, na qual ocorre como um grupo protético portador de ferro, bem como de numerosos citocromos, que estão envolvidos no metabolismo energético em praticamente todas as células da cadeia respiratória. Para a rotação contínua destas proteínas, a hemorragia deve ser constantemente fornecida.
Oito enzimas estão envolvidas na biossíntese da hemorragia; a regulação da síntese ocorre através da inibição do feedback da síntese δ-ALA, a primeira enzima da biossíntese, pela hemorragia do produto final. Existem duas isoenzimas no organismo: δ-ALA synthase 1 ocorre em todos os tecidos, δ-ALA synthase 2 apenas na medula óssea. Devido à sua importância quantitativa, o fígado e a medula óssea são os locais mais importantes para a síntese da hemorragia.
Fisiopatologia das porfírias
Mutações hereditárias (ou raramente espontâneas) nos genes das enzimas da biossíntese de hemoglobina levam a uma perda parcial da função da enzima codificada. Esta perda parcial da função (i.a. a 50%) é suficientemente compensada pela estimulação das sínteses δ-ALA, de modo a que normalmente não haja deficiência de hemorragia. Os portadores da predisposição genética geralmente não têm, portanto, qualquer problema com a redução do desempenho metabólico da enzima afectada. A situação torna-se problemática quando a necessidade de hemorragia aumenta, por exemplo, ao tomar uma droga como a fenitoína, que induz enzimas de degradação do citocromo P450 no fígado que contém hemorragia. A síntese da hemorragia é então muito fortemente estimulada e ocorre um “engarrafamento” no ponto estreito do defeito enzimático devido à redução da actividade para 50%: Os intermediários do metabolismo da hemoglobina (especialmente os precursores da porfirina porphobilinogénio e o δ-aminolevulinato) acumulam-se nas células, entram no espaço extracelular e distribuem-se no organismo.
Três formas de porfíria
Dependendo da enzima afectada nas porfírias agudas, é feita uma distinção entre a variante frequente “porfíria aguda intermitente” (AIP) e as variantes mais raras “porfíria variegata” (PV) e “coproporfirinúria hereditária” (HKP) (Fig. 1).
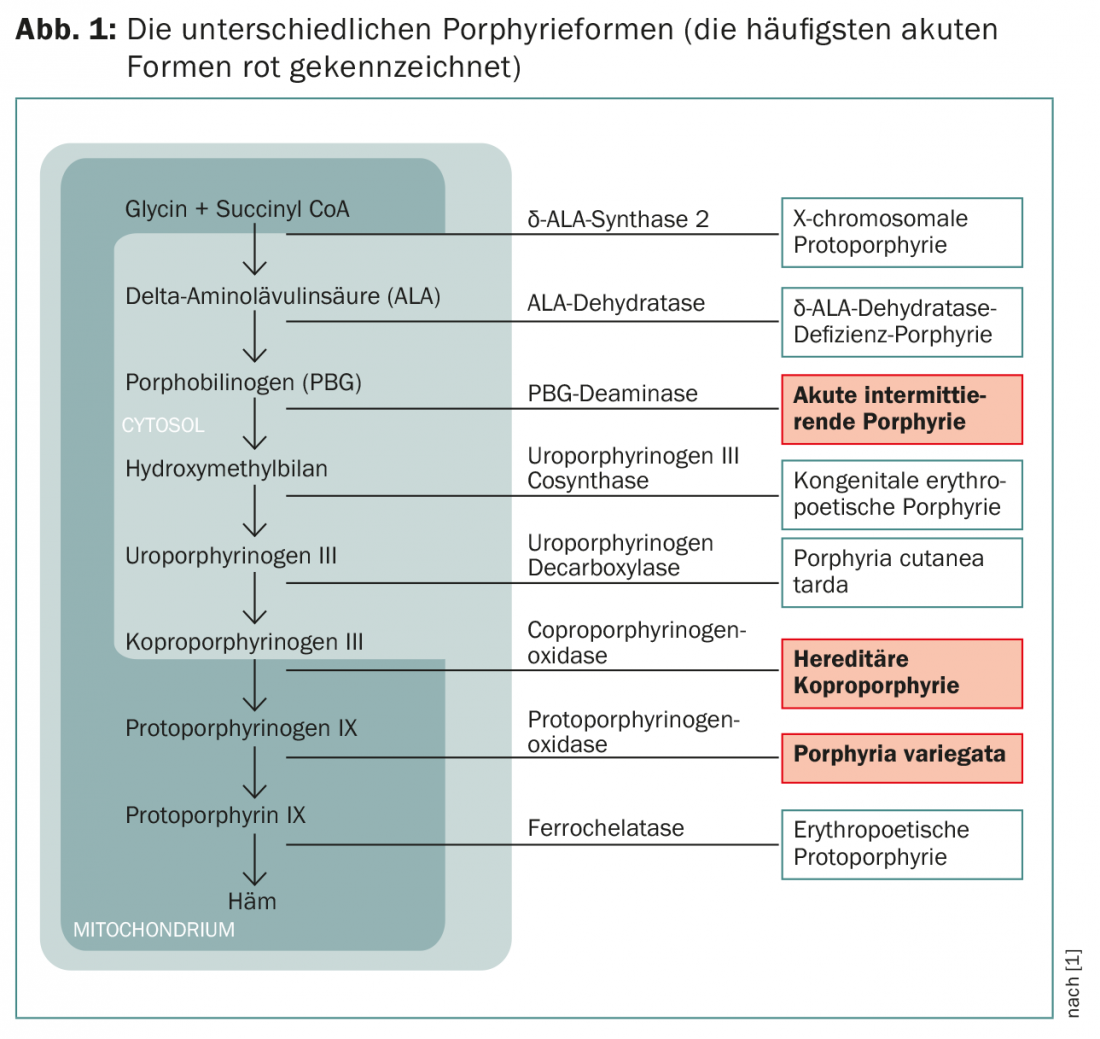
Os sintomas dependem de quais metabolitos se acumulam. Os precursores de porfirina, que ainda são solúveis em água, penetram nos nervos e causam sintomas neurológicos; as porfirinas solúveis em gordura são depositadas na pele e causam sintomas cutâneos através da exposição à radiação UV. Estas queixas são muito variadas e frequentemente confusas, uma vez que também ocorrem com um grande número de outras doenças. O diagnóstico correcto é, portanto, geralmente feito numa fase correspondentemente tardia.
Sintomas: Dores abdominais, urina vermelha, perturbações neurológicas
Nas três formas, a ocorrência aguda dos sintomas é chamada uma recaída. As recaídas podem ser leves e são então determinadas principalmente pela dor abdominal. Ocorrem repetidamente sem sucesso na realização do diagnóstico correcto. Só no caso de um episódio grave em que, para além da dor abdominal, ocorram sinais de sintomas centrais (tais como humor depressivo, alucinações, coma) e sintomas neurológicos periféricos (dor nas costas, paralisia) (tríade de sintomas), é que aumenta a probabilidade de ser feito o diagnóstico correcto (Fig. 2).
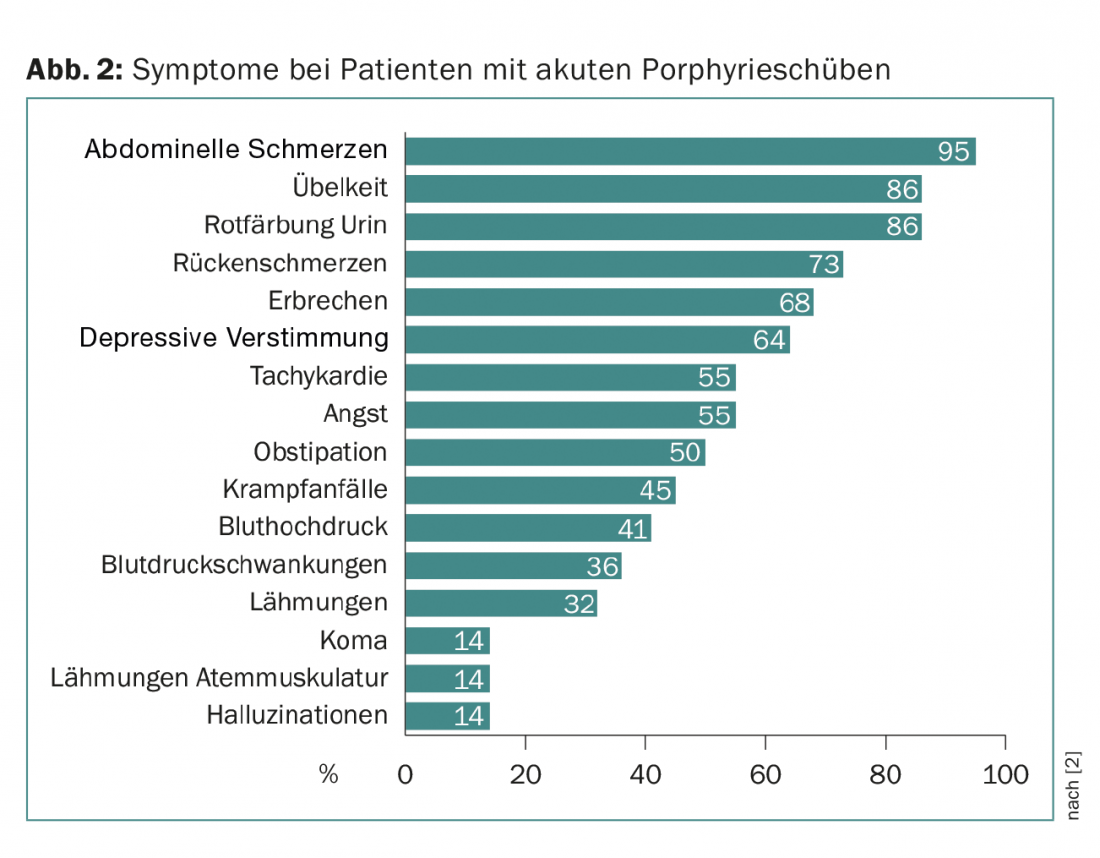
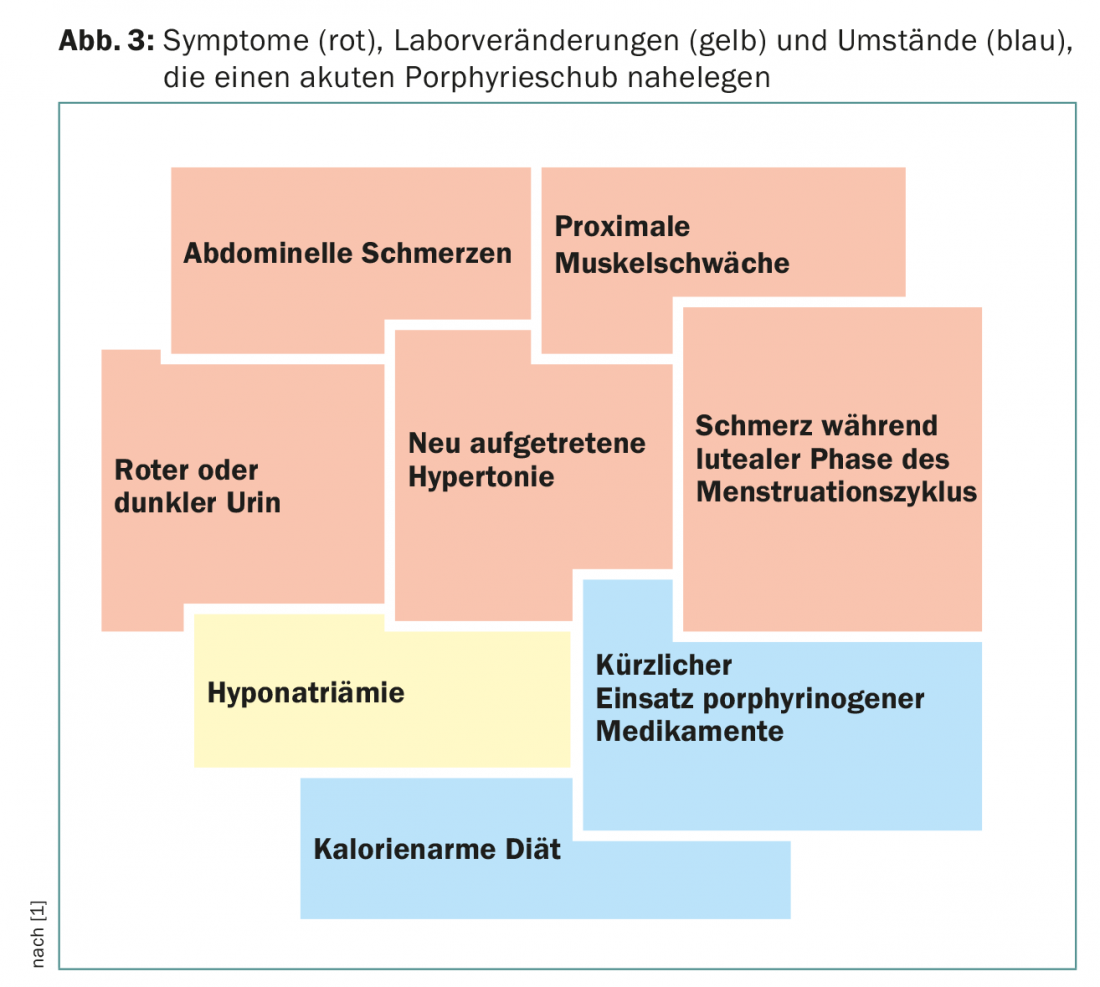
Um sinal clínico adicional importante é a descoloração vermelha da urina, que é causada pela auto-oxidação do porfobbilinogénio à porfobbilina. Juntamente com alterações laboratoriais tais como hiponatremia, estes sintomas sugerem porfíria aguda (Fig. 3). Os doentes são frequentemente tratados em unidades de cuidados intensivos devido à gravidade dos sintomas, e é frequentemente um jovem assistente que se lembra da doença rara devido à sua proximidade com o exame estatal.
A hiponatremia pode levar a convulsões, que não raro são tratadas com carbamazepina ou fenitoína, que estão entre as drogas que promovem as convulsões, sem conhecer o diagnóstico. Os sintomas cerebrais podem variar desde uma ligeira deficiência mental a psicose paranóica, delírio ou coma. Em casos individuais, ocorre mono ou hemiparesia central; em caso de paralisia respiratória, a necessidade de ventilação mecânica é iminente, para que se possa desenvolver uma situação de risco de vida. Na síndrome de encefalopatia reversível posterior (PRES), detectada por uma ressonância magnética, pode ocorrer cegueira cortical com perda total da acuidade visual.
Alguns doentes desenvolvem neuropatia periférica, predominantemente motora, durante a recidiva. Numa grande proporção das pessoas afectadas, a polineuropatia é desencadeada por medidas iatrogénicas (administração de fármacos porfirinogénicos), na ignorância da presença de porfíria aguda. A neuropatia é distribuída atipicamente, ou seja, os braços são mais severamente afectados do que as extremidades inferiores, os músculos proximais mais pronunciados do que os grupos musculares distais. Num terço dos pacientes, a paralisia começa nas pernas, ao meio, com ênfase proximal. A distribuição assimétrica da paresia é comum. Os sinais de neuropatia sensorial são relatados como sendo perturbações sensoriais, quer em forma de meias e luvas, quer na zona do tronco como um “fato de banho”. Nas porfírias agudas mais raras, PV e HKP, também ocorrem sintomas cutâneos. Isto deve-se ao facto de não só os precursores da porfirina serem elevados, mas também devido à posição do defeito enzimático responsável, produtos intermediários posteriores de síntese da porfirina são também depositados na pele.
Diagnósticos
A maior detecção de precursores de porfirina (δ-ALA e PBG) na urina e não – como ainda muitas vezes erroneamente se supõe – principalmente de porfirinas sugere a suspeita de porfíria aguda. O Porphobilinogénio é medido pela primeira vez de forma semi-quantitativa num teste rápido. O problema é que este teste não está disponível em muitos laboratórios. Se o teste for positivo, a análise quantitativa de PBG e de δ-ALA segue-se: se o resultado estiver relacionado com o valor de creatinina de urina, uma amostra espontânea de urina é geralmente suficiente. Já não é necessária urina 24 horas por dia.
Se os precursores da porfirina forem elevados, a presença de porfíria aguda intermitente é confirmada pela determinação da deaminase PBG nos eritrócitos e – se esta for reduzida – por uma análise de mutação (teste genético). Para o diagnóstico das outras duas porfírias agudas, é necessária a análise de urina e fezes para as uro- e coproporfirinas, respectivamente.
Actualmente, a confirmação final do diagnóstico é feita através de mais de 300 mutações diferentes no gene deaminase PBG, ou seja, praticamente cada segunda posição de aminoácidos pode ser afectada por uma mutação. Por conseguinte, o gene completo deve ser sempre sequenciado no diagnóstico inicial. Se a mutação for conhecida, é possível testar parentes especificamente para a presença desta mutação.
Apenas cerca de 10-15% dos portadores de genes desenvolvem uma manifestação de porfíria durante a sua vida. Isto é atribuído, por um lado, ao facto de um confronto com agentes desencadeantes não ocorrer no conhecimento da predisposição e, por outro lado, ao facto de os genes modificadores protegerem contra a manifestação de uma recaída.
Opções terapêuticas
Anteriormente, a única terapia disponível era a infusão de glicose de alta dose; isto inibe a síntese de hemorragia, regulando a actividade da síntase δ-ALA. Actualmente, a administração intravenosa de hemarginato (Normosang®) é a terapia de escolha para “abrandar” a biossíntese de hemorragia. A droga é administrada como uma infusão curta durante quatro dias; uma mistura com albumina humana é útil por causa do efeito irritante das veias. Os doentes individuais que sofrem de episódios de recaídas crónicas recebem infusões através de um port-a-cath. É evidente que os medicamentos promotores de recaídas devem ser descontinuados e não devem ser utilizados novamente. As drogas clássicas que provocam recaídas são, por exemplo, os anticonvulsivos fenitoína, carbamazepina, oxcarbazepina e ácido valpróico, barbitúricos, os analgésicos metamizol e fenilbutazona, os antibióticos sulfonamida e a droga antifúngica griseovulvina. Lorazepam, diazepam, morfina, petidina, clorpromazina e ondansetron, por exemplo, são considerados seguros.
Os pacientes recebem um cartão de emergência, que também se refere ao website www.drugs-porphyria.com, onde as listas de medicamentos recauchutados e seguros são constantemente actualizadas. Em casos individuais, é difícil realizar a atribuição com certeza.
Cuidados interdisciplinares
Actualmente, o diagnóstico e tratamento de doentes com porfírias agudas são efectuados numa equipa interdisciplinar com hematologistas, gastroenterologistas, neurologistas, anestesistas, médicos de laboratório, dermatologistas, ginecologistas e geneticistas [1]. Os seguintes aspectos são de particular importância:
- Recidivas crónicas recorrentes
- Tratamento de complicações neurológicas graves, relacionadas com recaídas
- Preparação e execução de anestesia
- Prevenção de diagnósticos errados
- Cuidados a pacientes grávidas com porfíria
- Diagnóstico diferencial de possíveis alterações cutâneas
- Fiabilidade da análise da mutação.
Novo diagnóstico: Inclusão no registo de porfíria
Devido à raridade das doenças, as informações sobre o curso, a gravidade das manifestações clínicas e a diminuição da qualidade de vida, possíveis efeitos secundários dos tratamentos, etc., só podem ser obtidas a partir de registos. No registo de Munique de porfírias agudas, 60 pacientes com porfírias agudas foram documentados a partir de Fevereiro de 2016. Os doentes recém-diagnosticados devem ser comunicados a este registo [2]. Está em construção um website sobre isto (www.akuteporphyrie.de).
Literatura:
- Petrides PE (ed.), et al: The Acute Porphyrias, um folheto informativo para médicos. 4ª edição 2015 Munique (disponível em Orphan Europe Ulm).
- Bronisch O, et al.: Acute Intermittent Porphyria in Germany: Interim Analysis of 45 Patients from a single institution (Munich). Congresso Internacional sobre Porfirinas e Porfírias, Dusseldorf, 9/2015.
InFo ONCOLOGY & HEMATOLOGY 2016; 4(2): 16-19