A leucemia linfoblástica aguda é o cancro infantil mais comum. É tratado de uma forma adaptada ao risco e é curável na maioria dos casos. Novos medicamentos inovadores como as imunoterapias estão em ensaios clínicos.
Com uma quota de cerca de 30% e 3,3 novos casos por 100.000 habitantes com menos de 15 anos de idade, a leucemia linfoblástica aguda (ALL) é o cancro infantil mais comum. O pico da idade é de cerca de cinco anos. Na Suíça, cerca de 50-60 crianças são diagnosticadas com TODAS todos os anos. O subtipo imunologicamente mais comum na infância é o B-precursor ALL, que se desenvolve a partir de células imaturas da série B do sistema linfático. TODOS os T-lymphoiesis ocorrem com menos frequência. Uma forma especial é a leucemia madura da célula B, que se baseia numa transformação maligna da célula B madura e é entendida como uma manifestação leucémica do linfoma de Burkitt. ALL é uma doença heterogénea caracterizada pela proliferação descontrolada de células progenitoras linfóides na medula óssea e no sangue periférico [1]. É agora considerada como uma doença que tem frequentemente grandes semelhanças morfológicas mas pode ter subentidades citogenéticas ou genéticas moleculares muito heterogéneas [2], o que também se correlaciona com uma resposta clínica heterogénea ao tratamento. Com técnicas modernas de sequenciação, a enorme heterogeneidade clonal desta doença pode ser demonstrada.
Causas
A causa do desenvolvimento da leucemia ainda não é clara. Os factores conhecidos mas que raramente ocorrem são a radiação ionizante e as síndromes congénitas. No entanto, isto explica menos de 10% de todas as doenças. As crianças com síndrome de Down têm um risco cerca de 20 vezes maior de desenvolver leucemia (ALL ou leucemia mieloblástica aguda) nos primeiros cinco anos de vida em comparação com as crianças não afectadas e saudáveis. Contudo, a mieloproliferação transitória ocorre ainda mais frequentemente (em 3-10%) nestas crianças em idade neonatal, o que pode ocasionalmente transformar-se mais tarde em leucemia. Outras alterações congénitas raras com risco aumentado de leucemia são a ataxia teleangiectásica, a síndrome de Fanconi e outras síndromes associadas a uma imunodeficiência ou aumento da fragilidade cromossómica. O facto de TODOS ocorrer mais frequentemente entre o segundo e o quinto ano de vida, de a doença ser mais comum nos países industrializados e a observação de que, no passado, a agregação ocorreu repetidamente, especialmente em regiões de novas aglomerações, levou a várias hipóteses associadas à infecção para o desenvolvimento da leucemia [3,4].
Sintomas
A proliferação de explosões leucémicas na medula óssea leva a um deslocamento da hematopoiese normal, o que explica os sintomas mais comuns, tais como palidez e fadiga devido a anemia ou tendência a sangramento devido a trombocitopenia. As infiltrações conduzem frequentemente a dores ósseas difusas e artropatias alternadas, que ocasionalmente se manifestam em crianças pequenas como relutância em mover-se ou mesmo recusa em andar. Além disso, pode ocorrer inchaço dos gânglios linfáticos e organomegalia.
Diagnósticos
No sangue, são frequentemente encontradas alterações em pelo menos duas séries de células sanguíneas, mais frequentemente trombocitopenia com anemia simultânea. A contagem de leucócitos pode ser normal, diminuída ou aumentada. A morfologia do hemograma fornece pistas de diagnóstico importantes, o diagnóstico final é feito por aspiração de medula óssea. Além de examinar a morfologia, o imunofenótipo das explosões leucémicas é determinado por meio da citometria de fluxo (FACS) e é realizada uma análise cromossómica. A imunofenotipagem permite determinar a fase de desenvolvimento do clone celular correspondente. O subtipo mais comum de leucemia nas crianças, o chamado “common-ALL”, caracteriza-se pela expressão dos marcadores de células B CD10 e CD19. A expressão de antigénios mielóides, geralmente sem significado prognóstico, pode ser detectada em até metade de TODOS os casos. Hoje em dia, os exames genéticos citogenéticos e moleculares estão a tornar-se cada vez mais importantes. É importante reconhecer os subgrupos mais importantes, uma vez que têm implicações terapêuticas. Por um lado, procuram-se alterações cromossómicas numéricas tais como hiperdiploidia ou hipodiploidia, bem como alterações estruturais tais como translocações, por exemplo t(12;21) (fusão dos genes ETV6/RUNX1) ou t(9;22) (fusão de BCR/ABL1), rearranjos MLL (MLL 11q23) e outras alterações.
Classicamente, a detecção destas alterações é feita com citogenética convencional (banda G) e/ou hibridação fluorescente in situ (FISH) nas células de leucemia. Nos últimos anos, a medição da doença residual mínima (DRM) da medula óssea foi estabelecida como parte do diagnóstico de acompanhamento para avaliar a resposta da leucemia ao tratamento. A resposta à terapia surgiu agora como um dos parâmetros prognósticos mais importantes. Actualmente, dois métodos principais são utilizados no diagnóstico da progressão, que se complementam na prática clínica diária. O método mais sensível é o controlo dos rearranjos dos receptores de imunoglobulina e de células T. Inicialmente, procuram-se rearranjos clonais específicos da leucemia, que são seguidos com PCR quantitativa em pontos de tempo terapêuticos específicos. O limite de detecção assim alcançado é aproximadamente uma célula de leucemia por 100.000 células normais da medula óssea. Uma técnica para a medição do MRD que é cerca de um nível de registo menos sensível baseia-se na monitorização do imunofenótipo associado à leucemia pela FACS. Uma sensibilidade de 0,001% pode ser alcançada [5].
Tratamento
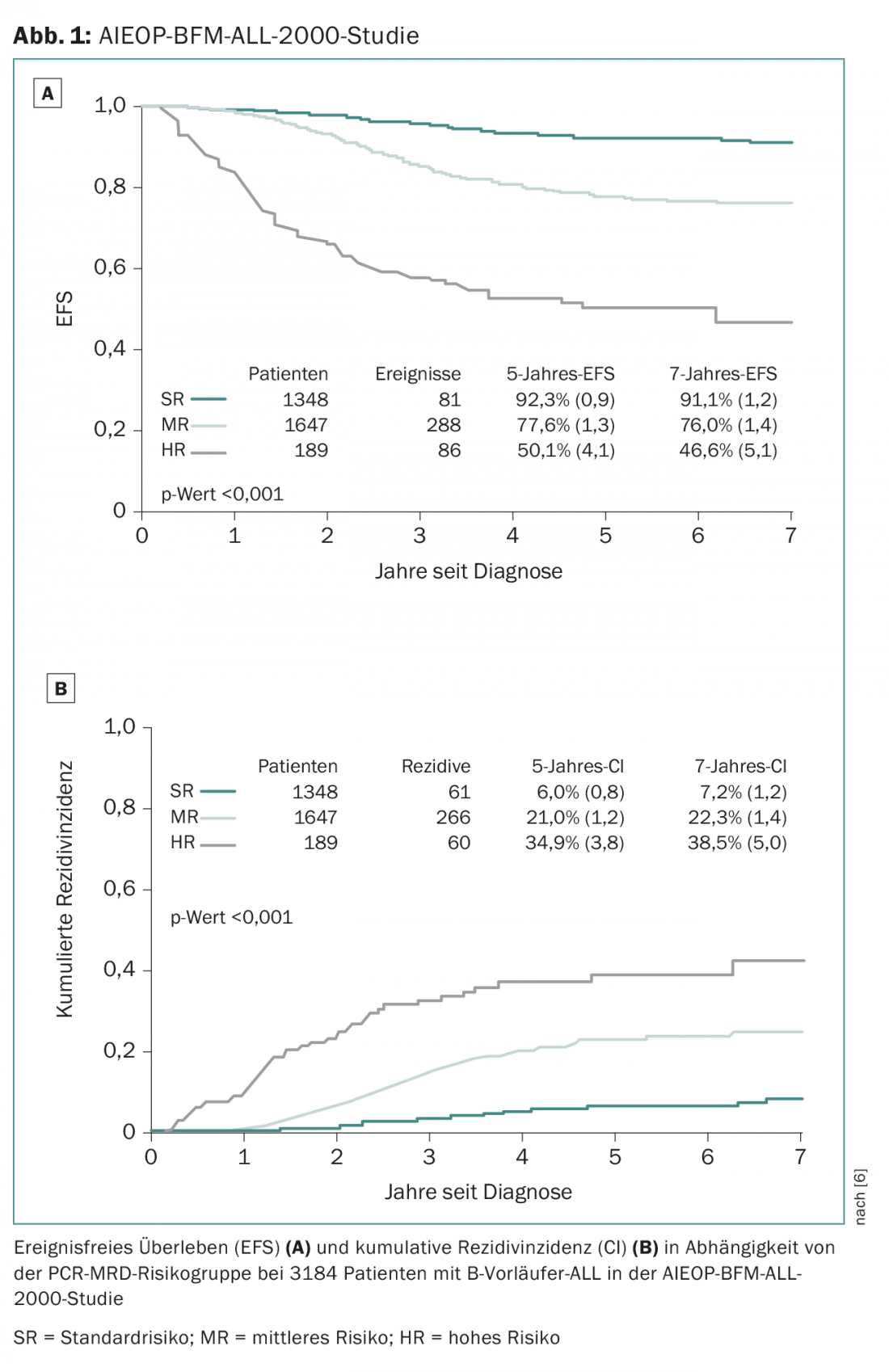
Nos anos 70, menos de 30% das crianças sobreviveram à doença, enquanto hoje em dia quase 85% dos doentes podem ser curados a longo prazo (Fig. 1) . O progresso nas taxas de sobrevivência nos últimos dez a 15 anos tem sido alcançado principalmente através de uma melhor estratificação do risco e do consequente tratamento adaptado ao risco. Actualmente TODAS as terapias da linha da frente consistem essencialmente na combinação de corticosteróides, um aminoácido ou esgotamento do substrato (asparaginase, metotrexato), substâncias alquilantes, antimetabolitos, bloqueadores clássicos de metafase e antraciclinas [6]. As substâncias mais recentes, as chamadas terapêuticas “específicas”, só têm sido utilizadas até agora de forma muito limitada no tratamento de TODOS pediátricos, com algumas excepções como os inibidores de tirosina quinase em Filadélfia cromossomo-positivo TODOS. Recentemente, foram identificados novos subgrupos raros de B-precursor ALL, os chamados “Philadelphia like” (ou “BCR-ABL like”), que por um lado mostram um risco significativamente aumentado de recorrência, mas por outro lado podem ser potenciais candidatos a terapias específicas [7]. Para a maioria dos novos critérios genéticos de alto risco, contudo, também poderia ser demonstrado que a sua influência varia, dependendo da resposta terapêutica mensurável.
A maioria dos centros oncológicos pediátricos suíços tratam os seus pacientes no âmbito dos estudos do grupo de estudo ALL-BFM, uma associação de centros oncológicos pediátricos alemães, austríacos e suíços, que tem contribuído significativamente para melhorias terapêuticas em TODOS em numerosos estudos terapêuticos randomizados de grande escala desde 1976. Após a maior parte do progresso nos ensaios anteriores ALL-BFM ter sido alcançado através de ajustamentos na classificação dos grupos de risco e na individualização do tratamento, estão a ser utilizados pela primeira vez novos medicamentos promissores no estudo de acompanhamento AIEOP-BFM ALL 2017, que está agora a ser planeado. A chamada “espinha dorsal” do tratamento é representada pelos medicamentos clássicos de TODOS os tratamentos acima mencionados. Além disso, os novos medicamentos inovadores são testados aleatoriamente para o seu potencial benefício. O estudo AIEOP-BFM-2017 previsto definirá novos grupos citogenéticos de alto risco (visão geral 1) que terão acesso a novas abordagens terapêuticas inovadoras. Um destes novos subgrupos é definido pela presença de uma eliminação IKZF1 em combinação com uma eliminação CDKN2A, CDKN2B, PAX5 ou PAR1 na ausência de uma eliminação ERG e é referido como IKZF1plus. Em estudos anteriores, cerca de 10-15% dos doentes puderam ser identificados como ICZF1plus e estes tinham uma taxa de recorrência significativamente mais elevada do que os doentes ICZF1plus-negativos [8]. Um destes novos medicamentos que será usado num pequeno grupo de grupos de alto risco com um prognóstico particularmente desfavorável é o blinatumomab, um anticorpo de célula T (BiTE) bi-específico que visa simultaneamente o receptor de célula T CD3 e a proteína de superfície de célula B CD19 [9]. O Blinatumomab destina-se a combinar dois efeitos potenciais: Redução de toxicidade aguda e a longo prazo, poupando a quimioterapia convencional e uma terapia mais eficaz dos pacientes que até agora só responderam de forma insatisfatória à terapia de alto risco.

Outro novo medicamento com um novo modo de acção em TODOS os tratamentos da linha de frente é o bortezomib inibidor do proteasoma. Desde tentativas anteriores de intensificação tardia da terapia em pacientes de alto risco tiveram pouco sucesso, e devido às já altas toxicidade das terapias de alto risco (AR), o bortezomib será randomizado para pacientes de AR na fase inicial pós-reindução no próximo ensaio.
Sistema nervoso central (SNC)
Actualmente, a prevenção de uma recidiva do SNC é feita principalmente com drogas, por um lado com injecções de metotrexato intratecal, por outro lado administrando drogas citostáticas de acção sistémica que se infiltram no cérebro (por exemplo, metotrexato de alta dose). Isto tornou possível limitar a radioterapia anterior do SNC, o que levou a uma redução dramática das recorrências do SNC mas foi associado a efeitos tardios não negligenciáveis, a situações de risco muito especiais [10,11].
Transplante de células estaminais
Após os resultados da terapia primária, bem como os protocolos de recaída das leucemias melhoraram significativamente ao longo do tempo, isto também levou a um ajustamento contínuo da indicação para terapias de altas doses com reinfusão de células estaminais. A indicação actual de transplante de células estaminais (SZT) como parte da terapia primária é reservada para certos subgrupos citogenéticos prognósticos desfavoráveis como o t(9;22), hipodiploidias com menos de 44 cromossomas nas explosões e IKZF1plus em combinação com uma resposta insuficiente à terapia (MRD) ao longo do tempo [12]. A experiência do grupo BFM mostrou que o sucesso do tratamento das recaídas depende do momento da ocorrência da recaída, do padrão da leucemia e do subtipo da leucemia [13]. No entanto, também foi aqui demonstrado que a resposta à terapia após a indução terapêutica renovada e, portanto, a dinâmica do declínio da doença residual mínima é de particular importância prognóstica e outros elementos terapêuticos, tais como a utilização de SCT, podem ser alinhados em conformidade [14].
Novas terapias
Com algumas excepções (clofarabina, nelarabina, imatinibe), não houve novas aprovações para TODOS os pediátricos nos últimos dez a 15 anos. No entanto, existem actualmente várias abordagens terapêuticas interessantes nos ensaios clínicos em estudos da fase I-III. Para além do cego acima mencionado, estes incluem também as células T do receptor quimérico de antígeno (CAR), com as quais TODAS as recidivas CD19-positivas já foram tratadas com sucesso. Esta é também uma imunoterapia que aproveita o potencial das células T citotóxicas autólogas para reconhecer e destruir as células da linha de células B. Outras imunoterapias promissoras, algumas delas associadas a citostáticos, estão actualmente em ensaios de fase I/II. Para além das imunoterapias, terapias orientadas após testes prévios in vitro em modelos de xenoenxertos ou linhas celulares ou inibidores específicos contra genes de fusão citogenéticos também representam opções terapêuticas interessantes e promissoras.
Mensagens Take-Home
- A leucemia linfoblástica aguda, o cancro infantil mais comum, é tratada de uma forma adaptada ao risco e tem cura na maioria dos casos.
- A determinação da doença residual mínima após indução terapêutica é um dos factores prognósticos mais importantes juntamente com marcadores biológicos, tais como o subtipo leucémico e alterações genéticas citogenéticas e moleculares nas explosões leucémicas.
- Os desenvolvimentos actuais visam um tratamento mais eficaz e direccionado dos subtipos de leucemia anteriormente resistentes, bem como uma redução da toxicidade da terapia.
- Novos medicamentos inovadores, tais como imunoterapias e abordagens terapêuticas individualizadas, estão em ensaios clínicos.
Literatura:
- Jabbour E, et al: Novos conhecimentos sobre a fisiopatologia e terapia da leucemia linfoblástica aguda em adultos. Cancro 2015; 121(15): 2517-2528.
- Pui CH, et al: Biologia, estratificação de risco, e terapia de leucemias agudas pediátricas: uma actualização. J Clin Oncol 2011; 29(5): 551-565.
- Kinlen L, et al: Infecções e factores imunitários no cancro: o papel da epidemiologia. Oncogene 2004; 23: 60-75.
- Greaves M, et al: Infecção, respostas imunitárias e a etiologia da leucemia infantil. Nat Rev Cancer 2006; 6(3): 193-203.
- Campano D, et al: Monitorização de doenças residuais mínimas na leucemia linfoblástica aguda infantil. Curr Opinião Hematol 2012; 19: 313-318.
- Conter V, et al: A resposta molecular ao tratamento redefine todos os factores prognósticos em crianças e adolescentes com leucemia linfoblástica aguda precursora de células B: resultados em 3184 pacientes do estudo AIEOP-BFM ALL 2000. Sangue 2010; 115(16): 3206-3214.
- Loh ML, et al: Tyrosine kinome sequencing of pediatric acute lymphoblastic leukemia: a report from the Children’s Oncology Group TARGET Project. Sangue 2013; 121(3): 485-488.
- Hinze L, et al: Impacto prognóstico das supressões IKZF1 em associação com pulsos de vincristina-dexametasona durante o tratamento de manutenção da leucemia linfoblástica aguda infantil no ensaio ALL-BFM 95. Leucemia 2017; 31: 1840-1842.
- Brentjens RJ, et al.: Segurança e persistência de células T autólogas CD19 transferidas adoptivamente em doentes com leucemias de células B refractárias recaídas ou quimioterápicas. Sangue 2011; 118(18): 4817-4828.
- Möricke A, et al.: A terapia ajustada ao risco de leucemia linfoblástica aguda pode diminuir a carga do tratamento e melhorar a sobrevivência: resultados do tratamento de 2169 pacientes pediátricos e adolescentes não seleccionados inscritos no ensaio ALL-BFM 95. Sangue 2008; 111(9): 4477-4489.
- Kamps WA, et al: tratamento orientado para BFM para crianças com leucemia linfoblástica aguda sem irradiação craniana e redução do tratamento para doentes de risco padrão: resultados do protocolo DCLSG ALL-8 (1991-1996). Leucemia 2002; 16(6): 1099-1111.
- Balduzzi A, et al: Quimioterapia versus transplante alogénico para a leucemia linfoblástica aguda infantil de muito alto risco na primeira remissão completa: comparação por randomização genética num estudo prospectivo internacional. Lancet 2005; 366: 635-642.
- Tallen G, et al: Resultado a longo prazo em crianças com recaída de leucemia linfoblástica aguda após estratificação em tempo e local de colapso e intensificação da quimioterapia multi-droga de curta duração: resultados do ensaio ALL-REZ BFM 90. J Clin Oncol 2010; 28: 2339-2347.
- Eckert C, et al: Minimal residual disease after induction is the strongest predictor of prognosis in intermediate risk recurpsed lymphoblastic leukaemia – Long-term results of trial ALL-REZ BFM P95/96. Eur J Cancer 2013 Apr; 49(6): 1346-1355.
InFo ONCOLOGy & HEMATOLOGy 2017; 5(5): 30-33