Se a formação de novas células sanguíneas na medula óssea for perturbada, a causa pode ser uma forma rara de cancro crónico do sangue. A hiperproliferação das três séries celulares na medula óssea causa eritrocitose, trombocitose e leucocitose na policitemia vera. O resultado é, entre outras coisas, um aumento significativo dos níveis de hematócrito e, portanto, também do risco de eventos tromboembólicos.
A policitemia vera (PV) é uma neoplasia mieloproliferativa crónica muito rara e caracteriza-se por um aumento da hematopoiese. A maioria dos doentes com PV tem uma mutação no gene JAK2 tirosina quinase [1]. Resulta num aumento da proliferação celular, bem como no aumento da produção de citocinas pró-inflamatórias. A sobreprodução de eritrócitos e o consequente aumento do hematócrito aumentam a viscosidade do sangue. Desta forma, a ocorrência de tromboembolismo é favorecida: 45% de todas as mortes em PV são devidas a complicações tromboembólicas [2]. No entanto, o prognóstico é geralmente favorável. A idade média do diagnóstico é de 65 anos.
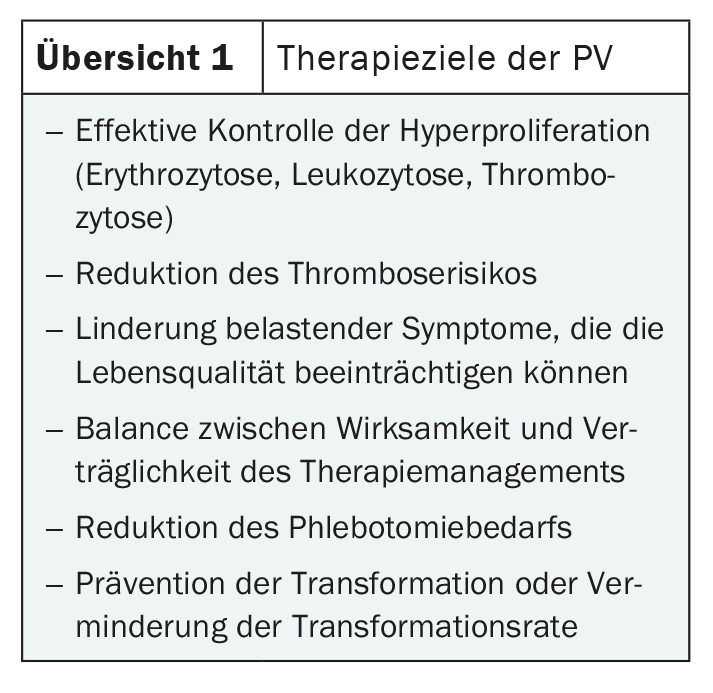
Embora os sintomas sejam variados, são geralmente pouco específicos. Por conseguinte, o diagnóstico é muitas vezes feito apenas por acaso. Os sintomas possíveis incluem dores de cabeça, distúrbios visuais, fadiga e prurido, bem como dores ósseas e dores na parte superior do abdómen. Estas são frequentemente causadas pela esplenomegalia típica da PV [1,2]. O aumento da massa de células sanguíneas pode causar distúrbios circulatórios que podem levar a tromboses venosas e arteriais graves, como embolia pulmonar, apoplexia ou enfarte do miocárdio. O diagnóstico precoce e o tratamento eficaz são, por conseguinte, indicados (Panorama 1) [3].
Na fase crónica, que geralmente dura anos, as características clínicas de mieloproliferação aumentada assumem um papel central. As complicações mais frequentes e potencialmente ameaçadoras são o tromboembolismo arterial ou venoso em até 40% dos doentes. Na fase tardia da doença, o principal problema reside na chamada fase “retardada”. Caracteriza-se por uma diminuição da eritrocitose e um aumento da esplenomegalia, combinados com fibrose da medula óssea, que pode ser seguida de transformação em mielofibrose (secundária) pós-PV e/ou leucemia aguda [1]. A taxa global de FPM pós-PV é de cerca de 15% após um período médio de observação de 10 anos e de 50% após 20 anos.
Tratamento adaptado ao risco
Uma vez que a prevenção do tromboembolismo é primordial, a flebotomia é muitas vezes considerada o tratamento de escolha. Isto pode baixar o hematócrito (Hct) para menos de 45% e reduzir a hiperviscosidade do sangue. Estudos demonstraram que a fixação de Hct abaixo de 45% pode reduzir a taxa de morte cardiovascular em PV para um quarto [4]. No entanto, uma flebotomia é muito extenuante. Por conseguinte, também deve ser iniciado um tratamento inicial com ácido acetilsalicílico (AAS) em dose baixa. A recomendação de tratamento é então baseada na pontuação de risco. Pode assumir-se um risco baixo para os doentes mais jovens, com menos de 60 anos de idade, que não tenham tido anteriormente uma trombose. Atualmente, está a ser discutido se o tratamento de redução de citocinas também deve ser considerado para eles em determinadas condições.

No entanto, a maioria dos doentes com PV apresenta um risco elevado. Nestes casos, é indicado o início da terapia citoreducativa. Recomenda-se Hydroxyurea (HU) ou interferon alfa (INF) para tratamento primário [5]. No entanto, a HU, em particular, não é adequada para todos os doentes e pode causar efeitos secundários graves (Panorâmica 2) [6]. Para pacientes mais jovens que desejam ter filhos, a INF é, portanto, mais susceptível de ser utilizada. Se a terapia de primeira linha não for tolerada ou se os sintomas clínicos não regredirem o suficiente, o tratamento deve ser alterado. O inibidor JAK2 ruxolitinibe foi demonstrado em ensaios para controlar o aumento da mieloproliferação, sendo ao mesmo tempo bem tolerado [1]. Muitos sintomas associados ao PV, tais como fadiga e prurido, também desapareceram. Além disso, a maioria dos pacientes experimentou o efeito muito rapidamente, dentro das primeiras quatro semanas. O Busulfan pode ser utilizado como terapia alternativa em pacientes de idade avançada. Aqui, no entanto, o potencial leucemogénico está sempre em discussão, razão pela qual a substância só deve ser utilizada com contenção. O anagrelide pode ser considerado como um parceiro combinado para, por exemplo, a HU ou INF. Isto destina-se exclusivamente a reduzir a produção de plaquetas e pode funcionar como um suplemento se não forem alcançados resultados satisfatórios apenas com as outras substâncias.
Literatura:
- www.onkopedia.com/de/onkopedia/guidelines/polycythaemia-vera-pv/@@guideline/html/index.html (última chamada em 23/04/2024).
- Vannucchi AM, et al.: N Engl J Med 2015; 372: 426–435.
- Stein BL, Moliterno AR, Tiu RV: Polycythemia vera disease burden: contributing factors, impact on quality of life, andemerging treatment options. Ann Hematol 2014; 93: 1965–1976.
- Marchioli R, Finazzi G, Specchia G, et al.: Cardiovascular events and intensity of treatment in polycythemia vera. N Engl J Med 2013; 368: 22–33.
- Barbui T, Tefferi A, Vannucchi AM, et al.: Philadelphia chromosome-negative classical myeloproliferative neoplasms: revised management recommendations from European Leukemia Net. Leukemia 2018; 32: 1057–1069.
- Barosi G, Birgegard G, Finazzi G, et al.: A unified definition of clinical resistance and intolerance to hydroxycarbamide in polycythaemia vera and primary myelofibrosis: results of a European LeukemiaNet (ELN) consensus process. Br J Haematol 2010; 148: 961–963.
InFo ONKOLOGIE & HÄMATOLOGIE 2024; 12(2): 38









