
A síndrome de Fanconi-Bickel (FBS) ocorre devido a variantes no gene SLC2A2. O diagnóstico de uma doença genética rara pode demorar até 5-6 anos, e ainda mais em países de baixo e médio rendimento com recursos tecnológicos limitados. Médicos do Peru apresentaram o caso de uma criança de dois anos e meio com atraso de crescimento, hepatomegalia, acidose metabólica, hipofosfatemia, hipocaliemia e hiperlactatemia.
A síndrome de Fanconi-Bickel (OMIM #227810), uma doença hereditária autossómica recessiva (AR), caracteriza-se por uma combinação de doença hepática e renal causada por um defeito no transportador de glucose GLUT2 (gene SLC2A2). Isto leva à acumulação de glicogénio, à disfunção tubular renal proximal e a uma diminuição da utilização da glicose e da galactose.
O fenótipo inclui falta de ganho de peso, distensão abdominal, hepatomegalia, hipoglicemia em jejum, hiperglicemia pós-prandial, glicosúria, fosfatúria, aminoacidúria, poliúria, acidose metabólica, osteoporose, hipofosfatemia, raquitismo e presença de glicogénio na biopsia hepática ou renal. Em casos raros, foi observado carcinoma hepatocelular devido à ativação da via de sinalização Wnt. No entanto, também foram notificados casos de doentes com sintomas clínicos ligeiros, incluindo aqueles com glucosúria pura. O gene SLC2A2 (OMIM *138160) contém 11 exões e a sua proteína GLUT2 é constituída por 524 aminoácidos e está localizada na membrana celular, sendo expressa em hepatócitos, enterócitos, túbulos proximais renais, células beta pancreáticas, neurónios e astrócitos. As variantes patogénicas do SLC2A2 alteram a entrada e a saída de glicose nos hepatócitos e reduzem a secreção de insulina devido ao aumento da sensibilidade das células beta na fase pós-prandial.
Relato de caso de um menino de 2 anos e meio
Um menino de 2 anos e 7 meses, nascido e criado no Peru, da quinta gravidez de pais aparentados, apresentou-se à equipa do Dr. Hernán Abarca-Barriga do Instituto Nacional de Salud (INS) Peru devido a vómitos, diarreia, acidose metabólica, hipocaliemia e hiperlactatemia, hipoatividade e febre. Devido a estes sintomas, tinha sido hospitalizado anteriormente com 1 ano e 10 meses e 2 anos e 2 meses [1].
A criança nasceu com 3620 g de peso à nascença, 49 cm de altura e 34 cm de perímetro cefálico (percentil normal) e Apgar 8-9. Em termos de desenvolvimento psicomotor, conseguiu controlar a cabeça com um mês, sentou-se sem ajuda aos sete meses e andou com apoio com um ano e seis meses. Falou as suas primeiras palavras com um ano e cinco meses, disse duas palavras com dois anos e nove meses e mostrou um sorriso social com um ano. Com 1 ano e 8 meses de idade, foi avaliado por apresentar baixo peso e crescimento reduzido, diarreia crónica, febre e aumento do volume abdominal (Fig. 1).
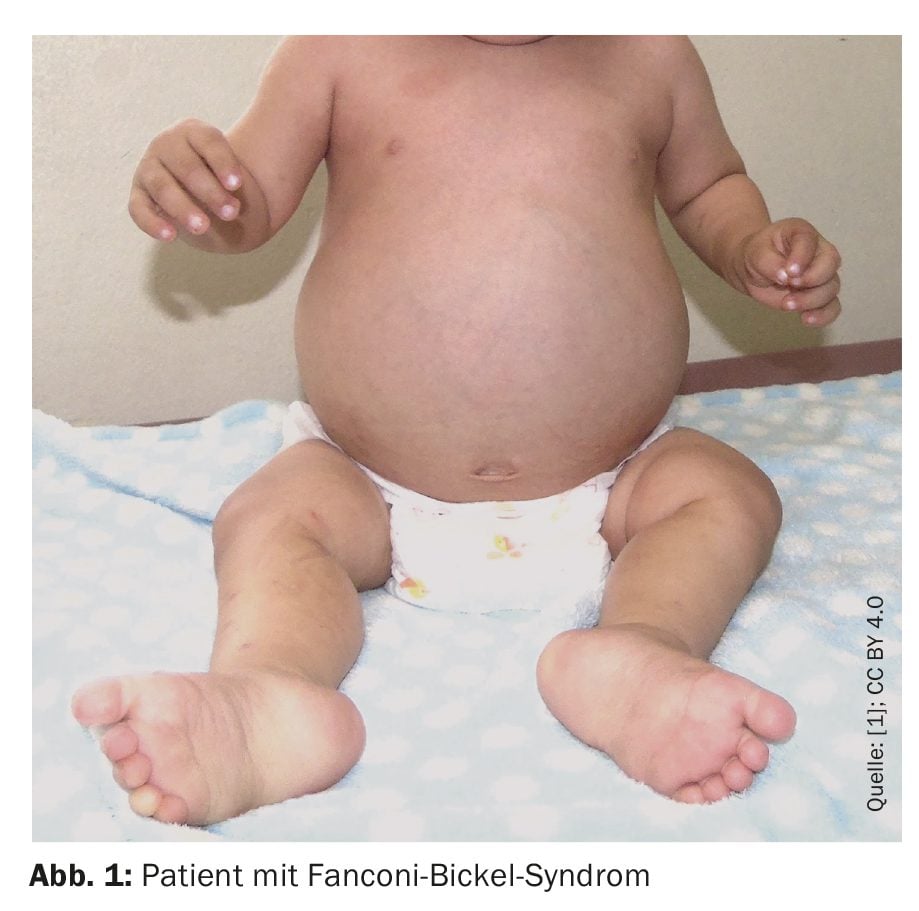
Aquando da admissão, os médicos diagnosticaram uma testa saliente, hepatomegalia, hipotonia e uma deformidade pseudo-madelung. O peso e a altura da criança estavam abaixo do primeiro percentil desde os seis meses de idade, enquanto o seu perímetro cefálico estava dentro da norma. As radiografias mostravam desgaste e alargamento das metáfises do fémur (distal) e da tíbia (proximal), consistentes com raquitismo.
Os resultados clínicos e laboratoriais sugerem que o FBS
O jovem doente apresentava hipoglicemia, hipo e hipercalcemia, hipofosfatemia, hipercolesterolemia, hipertrigliceridemia, hiperfosfatemia, hipocaliemia com diarreia e vómitos, hiperlactatemia e hipouricemia. A urinálise revelou valores normais de pH e HCO3, mas hiperproteinúria, hipocreatinúria, microalbuminúria, hiperglicosúria e hipercalciúria. O rapaz apresentava também trombocitose. A gasimetria venosa mostrava um pH de 7,261-7,5 mmHg, HCO3 de 7,5-28,8 mmHg e um excesso de bases de -15,8 a +5,3. Estas análises levaram ao diagnóstico de acidose tubular renal. A ecografia abdominal mostrava hepatomegalia e não havia sinais de fibrose ou nefromegalia.
A biópsia hepática revelou uma arquitetura hepática parcialmente distorcida devido à presença de algum alargamento fibroso do espaço portal, infiltrado inflamatório de linfócitos, hepatócitos grandes e em balão com um padrão em mosaico e fibrose pericelular ligeira. A coloração periódica de ácido-Schiff com diástase destaca depósitos eosinofílicos nos hepatócitos que se correlacionam com a deposição de glicogénio. Com base nestes achados clínicos e laboratoriais, os médicos suspeitaram da síndrome de Fanconi-Bickel (FBS).
A sequenciação do exoma identificou uma variante patogénica homozigótica
Os resultados dos testes genéticos foram obtidos com 1 ano e 8 meses de idade, utilizando ADN genómico. Foi identificado um total de 131.477 variantes anotadas em 18.179 genes, excluindo as variantes que eram provavelmente benignas ou ligeiras. Para identificar as variantes associadas ao fenótipo do doente, foram utilizados os termos “failure to thrive” (HPO: 0001508) e “hepatomegaly” (HPO: 0002240), tendo os autores referido que foi considerado um limiar de frequência alélica populacional de 1%. Devido a hipofosfatemia, raquitismo e armazenamento de glicogénio no fígado, os investigadores também procuraram manualmente variantes no SLC2A2. Devido à consanguinidade parental, foi dada prioridade à análise de homozigotos. Foi utilizada uma frequência de alelos variantes (VAF) superior a 0,9 para selecionar potenciais genes candidatos. A sequenciação do exoma identificou uma variante homozigótica sem sentido no gene SLC2A2, que foi descrita como patogénica.
O diagnóstico clínico do doente baseou-se na presença de hepatomegalia, hipoglicemia, glicosúria, hipofosfatemia, hiperfosfatemia, hipouricemia, raquitismo, deformidade pseudo-madelung e acidose tubular renal. No entanto, não foi possível determinar a presença de aminoacidúria, uma vez que o teste não estava disponível no local. Esta excreção insuficiente de aminoácidos teria facilitado o diagnóstico clínico-bioquímico, explicam os autores. A confirmação molecular utilizando a sequenciação Sanger do gene SLCA2 foi outro obstáculo, uma vez que este teste não está disponível no Peru. Dado o acesso limitado a testes genéticos no país, não foi possível confirmar a presença da variante nos familiares de primeiro grau do doente, sublinham os autores. A sequenciação do exoma permitiu aos investigadores identificar com precisão a variante homozigótica neste caso.
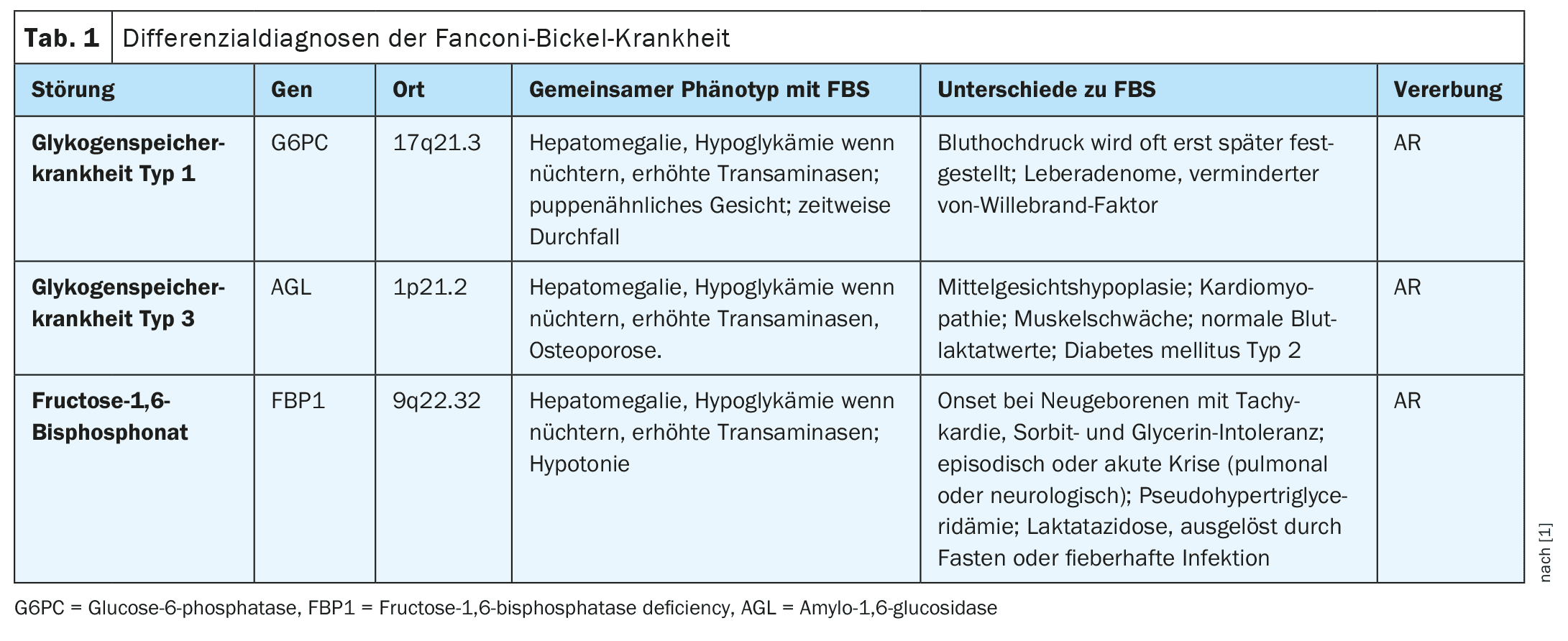
De acordo com o Dr. Abarca-Barriga e colegas, o aumento observado no lactato sanguíneo pode dever-se a uma carga acrescida de glucose proveniente do metabolismo anaeróbico. Além disso, um baixo nível de ácido úrico devido à disfunção dos túbulos proximais faz parte do fenótipo dos doentes com FBS, levando à hiperuricosúria. É provável que algumas caraterísticas clínicas, como o atraso no desenvolvimento da fala, também estejam associadas à presença de hipoglicemia crónica.
Atualmente, não existe uma terapia causal para a síndrome de Fanconi-Bickel. O jovem doente foi tratado com bicarbonato, amlodipina, citrato de sódio e solução de ácido cítrico, enalapril, alendronato e zolendronato e recebeu tratamento dietético com amido de milho não cozinhado, o que levou a uma melhoria do peso e da altura. Além disso, os autores sublinham que o doente apresentou uma melhoria de 1 desvio-padrão (DP) no peso e na altura desde o início do tratamento dietético com amido de milho não cozinhado. Por conseguinte, a utilização de amido de milho é essencial não só para prevenir a hipoglicemia nocturna, mas também para melhorar a altura e o peso do doente.
Literatura:
- Abarca-Barriga HH, et al.: Importance about use of high-throughput sequencing in pediatric: case report of a patient with Fanconi-Bickel syndrome. BMC Pediatr 2024; 24: 161; doi: 10.1186/s12887-024-04641-1.
HAUSARZT PRAXIS 2024; 19(11): 48–49









