
Para o prognóstico desta doença hereditária autossómica recessiva, o diagnóstico no período neonatal e o subsequente início do tratamento são cruciais – particularmente no que diz respeito à ocorrência de complicações, como carcinomas hepatocelulares e perturbações neurocognitivas. A nitisinona está atualmente disponível como terapia farmacológica, sendo também recomendadas certas medidas dietéticas.
Na tirosinemia hepatorenal de tipo 1 (HT1), a formação da enzima fumarilacetoacetase (FAH), a última enzima na degradação da tirosina, está comprometida desde o nascimento. Isto deve-se a uma variante patogénica bialélica no gene FAH. Devido à deficiência enzimática, os metabolitos tóxicos fumarylacetoacetate, maleylacetoacetate e succinylacetone acumulam-se no organismo e danificam o fígado, os rins e o sistema nervoso periférico. Como resultado, a insuficiência hepática não tratada ocorre frequentemente no primeiro ano de vida ou, num curso mais lento e crónico, desenvolve-se cirrose hepática ou carcinoma hepatocelular (CHC) [1]. A nitisinona é uma substância ativa que intervém na cascata de degradação precoce da tirosina, inibindo competitivamente a enzima 4-hidroxifenilpiruvato dioxigenase. Isto evita a formação de produtos intermédios tóxicos. O tratamento de todos os genótipos da doença deve ser iniciado o mais cedo possível, a fim de prolongar a sobrevivência global e evitar manifestações orgânicas potencialmente fatais. Na Suíça, a nitisinona (Nityr®) foi autorizada sob a forma de comprimidos em 2022 [2]. O início do tratamento nas primeiras semanas de vida não só previne o CHC na maioria dos casos, como também evita a disfunção hepática e renal [3–5].
Antes da era da nitisinona, a história natural da HT1 era geralmente fatal, 90% dos doentes com HT1 morriam nos primeiros dois anos de vida e a única opção era o transplante hepático [1]. A disponibilidade da nitisinona (NTBC) alterou assim fundamentalmente o curso clínico e o resultado das pessoas afectadas pela HT1, segundo os autores da diretriz S2k sobre o diagnóstico e o tratamento da tirosinemia hepatorenal (tirosinemia tipo 1), actualizada em 2022 [1].
Rastreio neonatal recomendado
O fator decisivo para o prognóstico a longo prazo, em particular para a prevenção do CHC, é o início do tratamento no período neonatal. Uma vez que os primeiros sintomas só aparecem na maioria dos doentes após vários meses de idade, o diagnóstico precoce não é possível por razões puramente clínicas [3]. Por conseguinte, em caso de suspeita clínica, a succinilazetona (SA) deve ser determinada quantitativamente a partir de sangue seco, soro/plasma e/ou urina (“rastreio seletivo”). A determinação isolada da concentração de tirosina tem uma sensibilidade e especificidade insuficientes, pelo que não é recomendada [1]. Um valor de medição de SA acima de um limite definido é considerado um resultado de despistagem positivo. Pode ser efectuada uma análise genética molecular do gene FAH para confirmar o diagnóstico (caixa). Se o gene FAHfor normal, as directrizes recomendam a análise do gene GSTZ1 [1].
| Análises de genética molecular A identificação de variantes patogénicas bialélicas no gene da fumarilacetoacetase (FAH)confirma o diagnóstico de tirosinemia tipo 1 [1]. O ADN das células sanguíneas ou de outros tecidos do corpo (esfregaço de bochecha) pode ser utilizado para esta análise. Também é possível efetuar análises de ARN a partir de células sanguíneas, amostras de biópsia hepática ou fibroblastos em cultura. A análise de um único gene dos 14 exões codificantes do gene FAHem doentes com um quadro clínico/bioquímico claro é um método há muito estabelecido que detecta variantes patogénicas com uma sensibilidade estimada de >95% [1]. Atualmente, porém, as técnicas de NGS(Next Generation Sequencing) são sobretudo utilizadas como parte de diagnósticos de painel ou como parte de WES (Whole Exome Sequencing) para a análise do gene FAH– ambos para diagnósticos mais rápidos e mais económicos [16]. |
Inicie a terapêutica com nitisinona nas primeiras semanas de vida
Os efeitos da nitisinona baseiam-se na inibição da enzima 4-hidroxifenilpiruvato dioxigenase, que está envolvida na degradação normal da tirosina (Fig. 1). Isto impede a formação de metabolitos tóxicos. Os efeitos secundários são raros, sendo os mais frequentemente comunicados os distúrbios da contagem sanguínea, os distúrbios oculares e o aumento da concentração de tirosina. A interrupção ou descontinuação abrupta da terapêutica com nitisinona pode levar a “crises de porfiria” e deve ser evitada [1, 6-8]. O valor-alvo terapêutico da nitisinona ainda não foi bem definido; na literatura especializada, 20-60 μM é dado como o valor-alvo terapêutico para as concentrações de nitisinona no plasma [3,9–11]. De acordo com os autores das orientações, são também possíveis concentrações significativamente mais baixas sem prejudicar o controlo metabólico, medido pela supressão da produção de SA [1]. O intervalo de objetivo terapêutico para a concentração de tirosina no sangue é indicado como sendo de 200-800 μM, sem que haja quaisquer estudos controlados, aleatórios e comparativos sobre este [3,9–12].
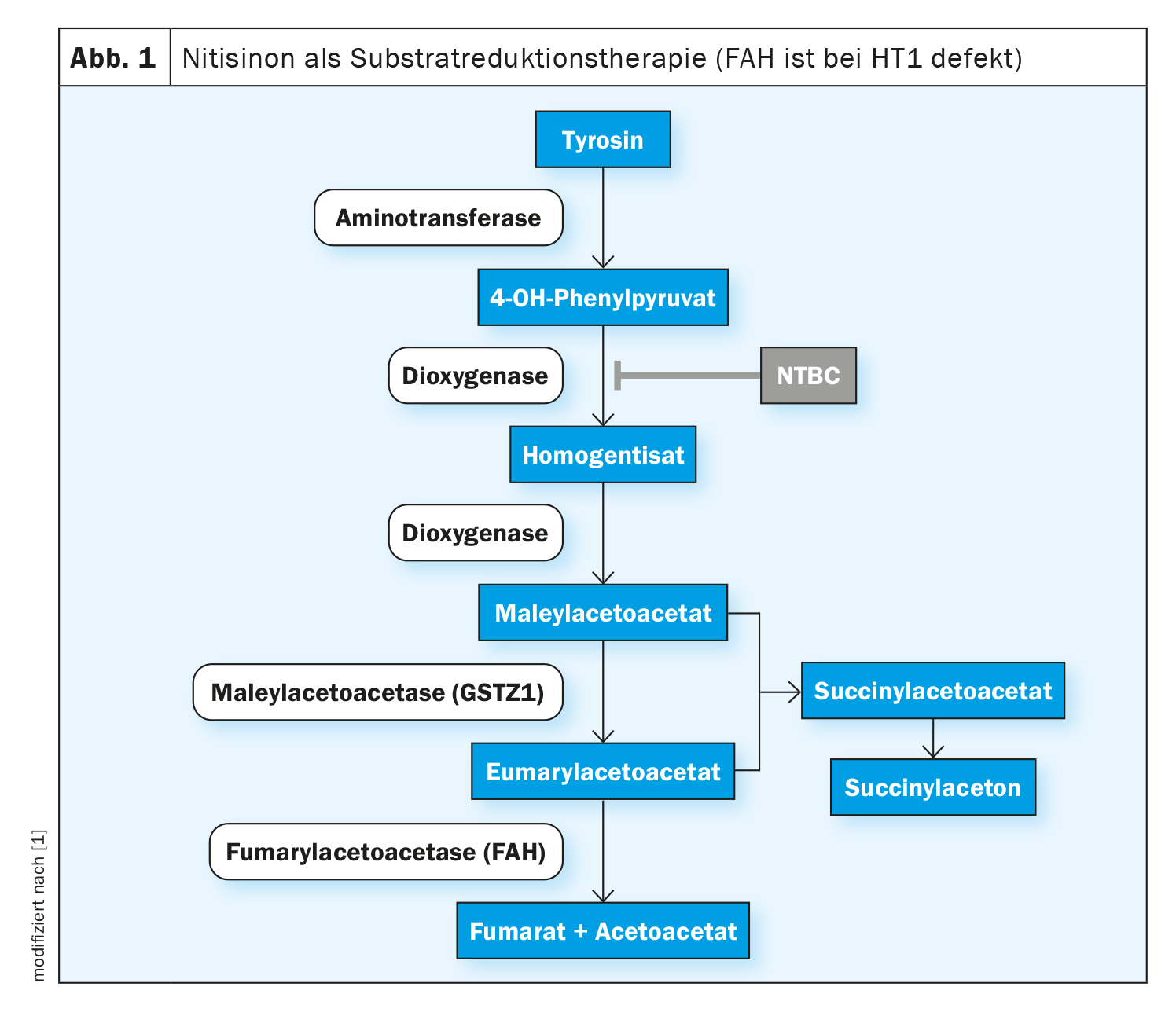
Se possível, a terapêutica com nitisinona deve ser combinada com uma dieta reduzida em proteínas suplementada com uma mistura de aminoácidos sem tirosina e fenilalanina [13–15].
Armadilhas de diagnóstico – as análises genéticas podem ajudar
O diagnóstico diferencial mais importante de uma concentração elevada de SA é a deficiência de maleil acetoacetato isomerase, uma anomalia metabólica presumivelmente benigna que não está associada a disfunção hepática [1]. A diretriz também salienta que concentrações ligeiramente elevadas de SA – dependendo do valor de corte utilizado – também podem ser temporárias ou devidas a formas ligeiras da doença que não requerem tratamento [1]. Um resultado de despistagem positivo deve ser confirmado por um ou mais métodos de análise alternativos, para além do controlo no mesmo material de amostra (sangue seco). Isto inclui a análise quantitativa de ácidos orgânicos na urina por cromatografia gasosa/espetrometria de massa (GC/MS) e a determinação de SA no sangue. Se houver uma forte suspeita, é também aconselhável verificar os parâmetros da função hepática. Os resultados anómalos no diagnóstico de confirmação podem ser esclarecidos através de testes genéticos moleculares do gene FAH. Se não for visível, pode ser analisado o gene GSTZ1 (glutationa S-transferase zeta 1-1), cuja deficiência é a causa da deficiência de maleil acetoacetato isomerase [1].
Literatura:
- “S2k guideline: Diagnosis and treatment of hepatorenal tyrosinaemia (tyrosinaemia type 1)”, número de registo AWMF: 027-003, a partir de 09.06.2022.
- Swissmedic: Informações sobre o medicamento, www.swissmedicinfo.ch,(último acesso em 06.02.2024)
- Mayorandan S, et al: Estudo transversal de 168 pacientes com tirosinemia hepatorenal e implicações para a prática clínica. Orphanet J Rare Dis 2014; 9: 107.
- McKiernan PJ, Preece MA, Chakrapani A: Outcome of children with hereditary tyrosinaemia following newborn screening (Resultados de crianças com tirosinemia hereditária após rastreio neonatal). Arch Dis Child 2015; 100(8): 738-741.
- Bartlett DC, et al: O tratamento precoce com nitisinona reduz a necessidade de transplante de fígado em crianças com tirosinemia tipo 1 e melhora a função renal pós-transplante. J Inherit Metab Dis 2014; 37(5): 745-752.
- Önenli Mungan N, et al: Tirosinemia tipo 1 e crise neurológica irreversível após um mês de interrupção da nitisona. Metab Brain Dis 2016; 31(5): 1181-1183.
- Schlump JU, et al: Crise neurológica grave num doente com tirosinemia hereditária tipo I após interrupção do tratamento com NTBC. J Inherit Metab Dis 2008; 31 Suppl 2: S223-S225.
- Uçar HK, et al: Relato de caso de uma associação muito rara de tirosinemia tipo I e pancreatite que imita uma crise neurológica de tirosinemia tipo I. Balkan Med J 2016; 33(3): 370-372.
- Chinsky JM, et al: Diagnóstico e tratamento da tirosinemia tipo I: uma revisão e recomendações do grupo de consenso dos EUA e do Canadá. Genet Med 2017; 19(12): doi:10.1038/gim.2017.101
- de Laet C, et al: Recomendações para a gestão da tirosinemia tipo 1. Orphanet J Rare Dis 2013; 8: 8. Publicado em 2013 Jan 11. doi:10.1186/1750-1172-8-8
- Alvarez F, Mitchell GA: Tirosinemia e transplante de fígado: experiência no CHU Sainte-Justine. Adv Exp Med Biol 2017; 959: 67-73.
- De Laet C, et al: Neuropsychological outcome of NTBC-treated patients with tyrosinaemia type 1. Dev Med Child Neurol 2011; 53(10): 962-964.
- Morrow G, Angileri F, Tanguay RM: Molecular Aspects of the FAH Mutations Involved in HT1 Disease (Aspectos Moleculares das Mutações FAH Envolvidas na Doença HT1). Adv Exp Med Biol 2017; 959: 25-48.
- Morrow G, Tanguay RM: Aspectos Bioquímicos e Clínicos da Tirosinemia Hereditária Tipo 1 Adv Exp Med Biol 2017; 959: 9-21.
- van Spronsen FJ, et al: Considerações dietéticas na tirosinemia tipo I. Adv Exp Med Biol 2017; 959: 197-204.
- Blackburn PR, et al: Tirosinemia silenciosa tipo I sem tirosina elevada ou succinilacetona associada a cirrose hepática e carcinoma hepatocelular. Hum Mutat 2016; 37(10): 1097-1105.
PRÁTICA DE CLÍNICA GERAL 2024; 19(2): 36-37









