A introdução de novos medicamentos melhorou constantemente o prognóstico dos doentes com mieloma múltiplo ao longo dos últimos anos. O diagnóstico também foi alargado, em particular através de análises citogenéticas, que permitem uma estratificação de risco mais precisa. No entanto, a taxa de sobrevivência de 5 anos na fase III é de apenas 40%, pelo que ainda há espaço para a inovação.
Sendo um linfoma de células B com proliferação monoclonal de células plasmáticas na medula óssea, o mieloma múltiplo (MM) é uma doença extremamente heterogénea. Enquanto cerca de um quarto das pessoas afectadas são assintomáticas no momento do diagnóstico, há também cursos agudos com rápida destruição óssea, disfunção renal pronunciada, fadiga, hipercalcemia e uma tendência para a infecção [1]. medida que a população envelhece, o número de novos casos aumenta, ocorrendo a maioria dos casos entre os 70 e 80 anos de idade. O mieloma múltiplo ainda é uma doença rara, com uma incidência de cerca de 6 casos por 100 000 habitantes em homens e 4 casos por 100 000 habitantes em mulheres, mas um aumento de um terço do número de casos pode ser esperado até 2040 só devido à mudança nas estruturas etárias [2,3]. Os requisitos para uma gestão eficiente com opções terapêuticas eficazes e diagnósticos orientados para alvos continuarão, portanto, a aumentar no futuro.
Um olhar sobre a fisiopatologia
No mieloma múltiplo, os plasmócitos malignos produzem imunoglobulinas monoclonais completas ou incompletas. Estas podem ser detectadas como cadeias de luz clonada aumentada em soro e urina e também ostentam o nome “paraproteína”. Na electroforese de proteínas séricas, aparecem sob a forma do famoso “gradiente M”. A elevada concentração destas imunoglobulinas pode causar vários sintomas, tais como amiloidose AL devido à deposição de proteínas desdobradas. Além disso, a síndrome de hiperviscosidade pode ocorrer e – muito mais frequentemente – a deterioração da função renal. Isto porque as cadeias de luz aumentadas são filtradas glomerularmente e muitas vezes não podem ser completamente reabsorvidas nos túbulos. Isto leva à excreção na urina, a chamada “Bence-Jones proteinuria”. Além disso, as paraproteínas acumulam-se nos glomérulos e precipitam-se no túbulo distal ligando-se à proteína Tamm-Horsfall produzida pelas células epiteliais tubulares. Descobertas: Glomerulosclerose e nefropatia de elenco [4].
Outros sintomas do mieloma múltiplo surgem principalmente do deslocamento da hematopoiese normal e da destruição dos ossos. Estes mecanismos levam a dores ósseas, fracturas patológicas, hipercalcemia, anemia e susceptibilidade à infecção, entre outras coisas [1]. Muitas vezes, as queixas das pessoas afectadas não são específicas, pelo que não é raro passarem vários meses antes de se fazer um diagnóstico correcto.
A genética do mieloma múltiplo é tão variada como a aparência clínica. As trissomias são encontradas em cerca de 40% dos pacientes. As translocações envolvendo a cadeia pesada de imunoglobulina (IgH) localizada no cromossoma 14q32 são também comuns. Juntamente com as trissomias, estas pertencem às alterações genéticas primárias e já podem ser detectadas nas fases preliminares da doença [5]. À medida que a doença progride, são acrescentadas aberrações genéticas secundárias, como as mutações del(1p) ou RAS, que moldam a clínica, a resposta à terapia e o prognóstico [3,6].
A conclusão é que a etiologia do mieloma múltiplo é pouco clara. Um aglomerado familiar é raro e os factores de risco permanecem pouco claros [3]. A doença desenvolve-se geralmente a partir de uma gamopatia monoclonal de significado desconhecido (MGUS) [7]. Se esta fase preliminar estiver presente, o risco de progressão para o mieloma múltiplo ou outra doença linfoproliferativa que requer tratamento é de cerca de 1% por ano [8]. No entanto, a detecção precoce dos precursores ainda não foi estabelecida, uma vez que isto ainda não levou a uma melhoria significativa no prognóstico [3].
Diagnósticos em transição
Actualmente, os critérios do Grupo de Trabalho Internacional sobre Mieloma M últiplo são utilizados principalmente para o diagnóstico do mieloma múltiplo (visão geral 1) [3,9]. A doença é classificada de acordo com o tipo de paraproteína, sendo os mielomas IgG e IgA os mais comuns. Se apenas se formam imunoglobulinas incompletas, isto é, cadeias ligeiras, fala-se de “mieloma de cadeia ligeira”. Estes representam cerca de um quinto dos casos [3].
Para além da anamnese, o exame físico, o hemograma e o laboratório, uma TAC de corpo inteiro de dose baixa e uma punção de medula óssea também fazem parte do diagnóstico inicial. As imagens são utilizadas para detectar osteólise e osteopenia [3]. Se necessário, isto pode ser prolongado por um exame de ressonância magnética para uma diferenciação mais precisa dos focos ósseos e para o diagnóstico de manifestações extramedulares. Isto pode ser particularmente útil para diferenciar do mieloma latente [10]. Os scans FDG-PET também podem fornecer informações sobre focos extramedulares e resposta terapêutica, mas actualmente não fazem parte das investigações padrão [11]. Se houver suspeita de envolvimento extramedular ou se estiverem presentes sintomas neurológicos, deve ser encomendada uma RM de regiões específicas, o mais tardar antes de se iniciar a terapia. A ecocardiografia também é indicada em caso de suspeita de amiloidose cardíaca [3].
Cada vez mais, os diagnósticos genéticos desempenham um papel importante na gestão do mieloma múltiplo. As análises em del(17p), t(4; 14) e t(14; 16) correspondem hoje ao work-up mínimo [3]. Estas alterações genéticas estão associadas a um prognóstico significativamente pior. Outras aberrações e também escores prognósticos baseados na expressão genética são prognósticos relevantes mas actualmente não (ainda) preditivos para terapias específicas [6,12].
Classificação para avaliar o prognóstico
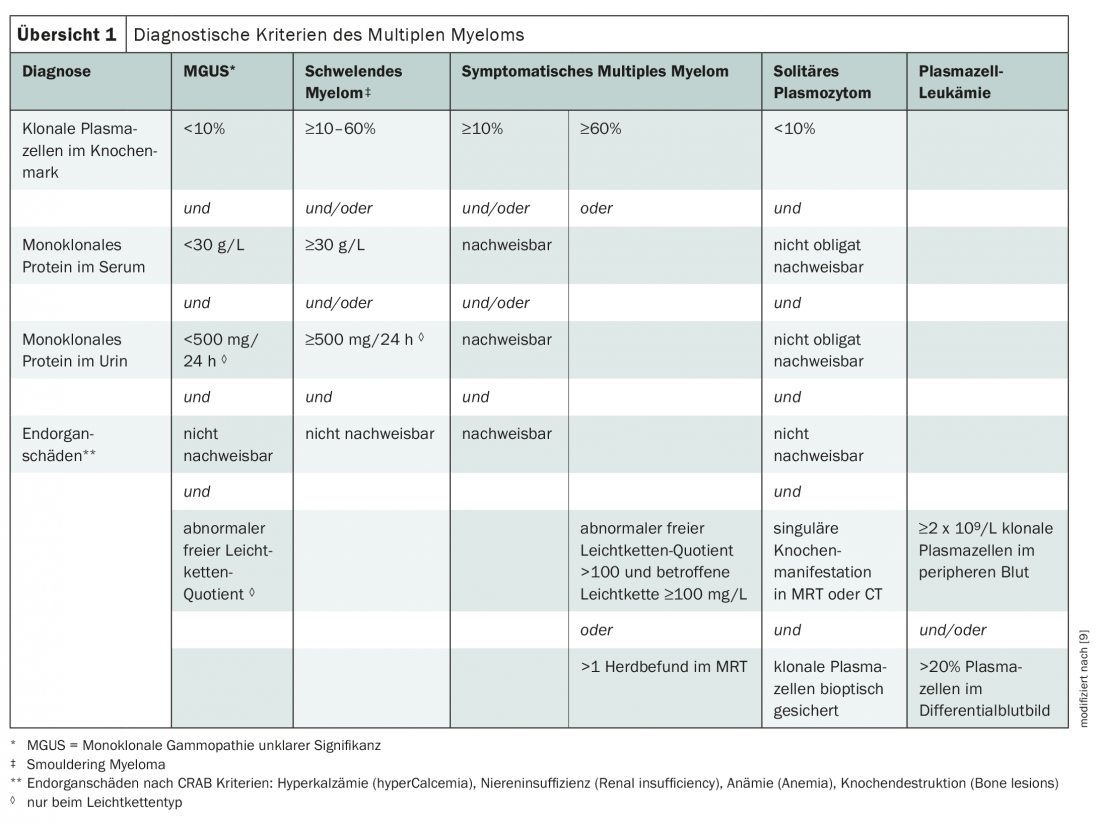
A encenação do mieloma múltiplo desempenha um papel importante na estimativa do prognóstico e na avaliação da terapia mais apropriada. A classificação segundo Salmon and Durie, que está em uso há muito tempo, é agora obsoleta. O Sistema Internacional Revisto de Encenação (R-ISS) do Grupo Internacional do Mieloma M últiplo (IMWG) é utilizado no seu lugar (Tab. 1) [12]. Com base nesta encenação, os indivíduos afectados são divididos em três grupos prognósticos, tendo em conta a albumina sérica, beta 2-microglobulina, LDH e aberrações citogénicas.
A presença de doença residual mínima (DRM) após a terapia é também prognosticamente significativa. Está presente na grande maioria dos doentes, mesmo depois de ter conseguido uma remissão completa de acordo com os critérios actuais e correlaciona-se com um resultado pior [13]. Uma vez que as análises do MRD ainda não têm qualquer valor preditivo para a continuação da terapia, não são actualmente um padrão nos exames de seguimento [3].
Terapia de primeira linha num relance
Para além dos cursos sintomáticos e dos chamados critérios CRAB (hipercalcemia – C, insuficiência renal – R, anemia – A e envolvimento ósseo – B), existem também outros parâmetros radiológicos e serológicos que, de acordo com os critérios do IMWG, representam indicações para o início da terapia. Estes incluem evidências de biomarcadores definidores de mieloma, um conteúdo de células plasmáticas clonadas na medula óssea de >60%, um quociente de cadeia de luz sem soro de >100 (cadeia de luz afectada/não afectada) e lesões focais de mais de 1 cm na ressonância magnética [3].
Se houver uma indicação de terapia, é feita uma distinção principalmente entre os pacientes que são adequados para transplante de células estaminais e aqueles que têm de ser tratados sem terapia de altas doses devido a comorbilidades. Se o transplante não for uma opção, várias combinações de duas e três drogas podem ser utilizadas no tratamento medicamentoso. Os doentes afectados com insuficiência renal, alta actividade da doença e citogenética desfavorável devem de preferência receber terapia com bortezomib como um componente [3]. Outros agentes que são utilizados são a ciclofosfamida, melfalan, lenalidomida e esteróides. Infelizmente, apenas alguns dos esquemas comuns foram directamente comparados. No geral, as combinações de três substâncias utilizando um inibidor proteasómico, um imunomodulador e um esteroide parecem ser superiores às combinações duplas. Se a condição geral for boa, estas são portanto preferíveis [3].
Se o transplante de células estaminais autólogas estiver disponível, é o tratamento de escolha na primeira linha de terapia. Até agora, nenhum novo medicamento foi capaz de igualar o transplante em termos de taxa de remissão, profundidade de resposta e sobrevivência sem progressão [14,15]. Os efeitos adversos da terapia de altas doses são um factor limitante para a indicação de transplante autólogo de células estaminais. Isto requer um bom funcionamento dos órgãos e a ausência de comorbidades significativas [16]. Há apenas alguns anos atrás, recomendava-se a terapia de indução de vincristina-antraciclina. Desde então, isto foi substituído por terapias combinadas com os novos medicamentos talidomida, bortezomibe e lenalidomida, o que leva a taxas de resposta significativamente melhores. Também aqui os dados sobre a comparação directa das diferentes terapias de combinação são infelizmente limitados; as remissões completas são atingidas em 20-40% dos casos [3]. Actualmente, a escolha da terapia de indução baseia-se principalmente em factores individuais do paciente, efeitos secundários e disponibilidade de medicamentos.
O transplante de células estaminais após terapia de indução pode ser realizado como um transplante único ou em tandem. Neste último caso, um segundo transplante autólogo é realizado no prazo de seis meses. Isto leva a um prolongamento da sobrevivência sem progressão e, nos doentes em fase R-ISS III, observou-se uma sobrevivência global mais longa, para além de uma sobrevivência sem progressão mais longa [17]. No entanto, o aumento da toxicidade de uma segunda terapia de alta dose também deve ser tido em conta. Actualmente, o transplante em tandem é recomendado para pacientes do grupo R-ISS III e para aqueles com citogenética de alto risco, tendo em conta vários subgrupos e análises a longo prazo [3]. Evidentemente, a presença de um número suficiente de células estaminais autólogas preservadas deve ser assegurada. Actualmente, o melphalan 200 mg/m2 é utilizado para a terapia de alta dose [18]. Os regimes de condicionamento alternativos que utilizam a ciclofosfamida e/ou irradiação corporal total já não são recomendados devido à sua maior toxicidade. A terapia óptima de alta dose também ainda é actualmente objecto de vários estudos; uma administração adicional de busulfan não levou a um prolongamento da sobrevivência global. Infelizmente, a adição de bortezomib também não se revelou eficaz [19]. Não existem dados conclusivos sobre a implementação da terapia de consolidação após transplante autólogo de células estaminais; isto pode ser particularmente útil em pacientes com resposta subótima após o transplante ou com citogenética de alto risco [20].
As recomendações para a terapia de manutenção são mais claras. Após anos sem ser recomendado, é agora uma parte importante do padrão terapêutico – graças aos novos medicamentos. Assim, o bortezomib ou lenalidomida é utilizado no grupo de alto risco e a lenalidomida no grupo de risco padrão. E este é também o caso com resposta completa [3,19,21].
Um factor terapêutico importante que não deve ser esquecido, independentemente do estatuto de transplante, é a osteoprotecção. Especialmente em casos de envolvimento ósseo e durante a terapia com glucocorticóides, os bisfosfonatos ou – especialmente em casos de insuficiência renal – denosumab devem ser utilizados para melhor prevenir a perda óssea. O zolendronato é considerado o bisfosfonato de primeira escolha para o mieloma múltiplo [3].
E em caso de progressão ou recidiva?
Progressões e recaídas após a terapia de primeira linha são um grande desafio no mieloma múltiplo. Não menos importante porque a população de doentes é ainda mais inomogénea com as linhas de terapia posteriores. As pessoas afectadas receberam tratamentos diferentes e tiveram experiências diferentes. A escolha dos medicamentos baseia-se, entre outras coisas, na eficácia e tolerabilidade do tratamento de primeira linha. Assim, se a experiência da primeira linha for boa, um medicamento da mesma classe de substância pode ser utilizado no tratamento da segunda linha. No entanto, em caso de baixa eficácia ou má tolerância, é indicada uma mudança de classe de fármacos. Actualmente, há uma abundância de novas drogas e combinações de drogas. Dependendo da apresentação clínica, das terapias e comorbilidades anteriores, uma vasta gama de substâncias e sequências pode assim ser seleccionada [3].
Embora as recaídas precoces e tardias sejam preferencialmente tratadas por transplante de células estaminais, existem vários regimes terapêuticos para todas as outras recaídas e para contra-indicações ao transplante. Estas são baseadas em inibidores proteasómicos ou imunomoduladores e consistem em duas a três substâncias activas (visão geral 2) . O anticorpo anti-CD38 daratumumab é também utilizado aqui. Em geral, as combinações triplas são mais eficazes do que as duplas combinações, mas também devem ser tidas em conta mais toxicidades [3,19].

Com a crescente investigação de novos agentes activos para a terapia do mieloma múltiplo, alguns sucessos foram alcançados nos últimos anos, particularmente na terapia de manutenção e em linhas de tratamento avançadas. No entanto, o prognóstico no grupo de alto risco e nas doenças recorrentes é ainda hoje desfavorável. Devido à análise citogenética cada vez mais generalizada, já é possível uma melhor estratificação do risco. Isto suscita esperanças de que no futuro, não só as opções terapêuticas de diagnóstico, mas também potencialmente mais direccionadas, estarão disponíveis.
Literatura:
- Kyle RA, et al: Revisão de 1027 doentes com mieloma múltiplo recentemente diagnosticado. Mayo Clin Proc. 2003; 78(1): 21-33.
- NICER: Estatísticas nacionais sobre a incidência do cancro. (último acesso: 27.02.2021)
- Wörmann B, et al: Mieloma múltiplo. Directriz Onkopedia. www.onkopedia.com/de/onkopedia/guidelines/multiples-myelom/@@guideline/html/index.html (último acesso 27.02.2021)
- Dimopoulos MA, et al: Patogénese e tratamento da insuficiência renal no mieloma múltiplo. Leucemia. 2008; 22(8): 1485-1493.
- Mikulasova A, et al.: O espectro das mutações somáticas na gamopatia monoclonal de significado indeterminado indica uma paisagem genómica menos complexa do que a do mieloma múltiplo. Haematologica. 2017; 102(9): 1617-1625.
- Bergsagel PL, Chesi MV: Classificação molecular e estratificação de risco do mieloma múltiplo. Hematol Oncol. 2013; 31 Suppl 1(0 1): 38-41.
- Landgren O, et al.: A gamopatia monoclonal de significado indeterminado (MGUS) precede consistentemente o mieloma múltiplo: um estudo prospectivo. Sangue. 2009; 113(22): 5412-5417.
- Dispenzieri A, et al: Prevalência e risco de progressão da gamopatia monoclonal da cadeia de luz de significado indeterminado: um estudo de coorte retrospectivo baseado na população. Lanceta. 2010; 375(9727): 1721-1728.
- Rajkumar SV, et al: International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014; 15(12): e538-548.
- Dimopoulos MA, et al: Papel da ressonância magnética na gestão de doentes com mieloma múltiplo: uma declaração de consenso. J Clin Oncol. 2015; 33(6): 657-664.
- Cavo M, et al: Papel do (18)F-FDG PET/CT no diagnóstico e gestão do mieloma múltiplo e outras doenças de células plasmáticas: uma declaração de consenso do Grupo de Trabalho Internacional sobre Mieloma Múltiplo. Lancet Oncol. 2017; 18(4): e206-e17.
- Palumbo A, et al: Revised International Staging System for Multiple Myeloma: A Report From International Myeloma Working Group. J Clin Oncol. 2015; 33(26): 2863-2869.
- Munshi NC, et al: Association of Minimal Residual Disease With Superior Survival Outcomes in Patients With Multiple Myeloma: A Meta-analysis. JAMA Oncol. 2017; 3(1): 28-35.
- Barlogie B, et al.: Superioridade do transplante autólogo tandem em relação à terapia padrão para mieloma múltiplo previamente não tratado. Sangue. 1997; 89(3): 789-793.
- Gay F, et al: Quimioterapia mais lenalidomida versus transplante autólogo, seguido de lenalidomida mais prednisona versus manutenção de lenalidomida, em doentes com mieloma múltiplo: um ensaio aleatório, multicêntrico, fase 3. Lancet Oncol. 2015; 16(16): 1617-1629.
- Merz M, et al: Sobrevivência de doentes idosos com mieloma múltiplo – Efeito do transplante inicial de células estaminais autólogas. Eur J Cancro. 2016; 62: 1-8.
- Cavo M, et al: Duplo Transplante Autólogo de Células-Tronco Prolonga Significativamente a Sobrevivência Sem Progressão e a Sobrevivência Global em Comparação com um Único Autotransplante em Mieloma Múltiplo Recentemente Diagnosticado: Uma Análise do Estudo EMN02/HO95 da Fase 3. Sangue. 2017; 130 (suplemento 1): 401.
- Giralt S: 200 mg/m(2) melphalan – o padrão de ouro para mieloma múltiplo. Nat Rev Clin Oncol. 2010; 7: 490-491.
- Dimopoulos MA, et al: Mieloma múltiplo: EHA-ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Anais de Oncologia. 2021; 32(3): 309-322.
- Einsele H, et al: Consolidação adaptada à resposta com bortezomib após ASCT melhora a sobrevivência sem progressão em mieloma múltiplo recentemente diagnosticado. Leucemia. 2017; 31(6): 1463-1466.
- McCarthy PL, et al: Manutenção da lenalidomida após transplante autólogo de células estaminais em mieloma múltiplo recém-diagnosticado: Uma meta-análise. J Clin Oncol. 2017; 35(29): 3279-3289.
InFo ONCOLOGy & HEMATOLOGy 2021; 9(2): 22-26









