As melhorias na gestão de doenças aumentam a qualidade de vida e a esperança de vida dos doentes com fibrose cística. O rastreio do recém-nascido introduzido em 2011 permite o diagnóstico precoce. Contudo, a doença deve também ser considerada como um possível diagnóstico diferencial na idade adulta.
A fibrose cística (FC, por vezes também fibrose cística) é a doença metabólica congénita mais comum, que acaba por limitar a vida na Suíça. Sendo uma doença sistémica, a FC afecta principalmente a função das glândulas exócrinas do tracto respiratório e do tracto digestivo. Em particular, o curso pulmonar com contínua destruição irreversível do órgão causa a diminuição da esperança de vida. Devido à melhoria das medidas médicas, os afectados atingem agora a idade adulta quase sem excepção, mas continuam a representar um colectivo relativamente novo na prática dos médicos de clínica geral. Como uma inovação promissora, uma terapia causal está disponível para uma proporção muito pequena de pacientes desde o início de 2014.
Padrão da doença
Já em 1936, a CF, que nessa altura ainda tinha um nome diferente, foi descrita pelo pediatra de Zurique Guido Fanconi como uma doença fatal de crianças pequenas, razão pela qual o acesso a ela permaneceu fechado aos colegas de medicina familiar durante muito tempo.
A FC é a doença congénita crónica mais comum e, em última análise, hereditária limitadora da vida, com uma prevalência de aproximadamente 1:2500. Aproximadamente a cada 25ª pessoa na Europa Central é um portador de mutação saudável. Existem cerca de 70.000 pessoas com a doença em todo o mundo, e cerca de 900 na Suíça. Cerca de metade dos doentes têm mais de 18 anos de idade. O modo de herança é autossómico recessivo, o que significa que estatisticamente uma em cada quatro crianças nascidas de uma relação entre dois portadores de mutação saudáveis é afectada pela FC, duas crianças são também portadores de mutação saudáveis como os seus pais, e uma criança não é portadora de mutação nem está doente. Em 1989, o gene subjacente foi identificado no cromossoma 7. A causa é uma mutação no gene CFTR (“cystic fibrosis transmembrane conductance regulator”), que codifica o canal do cloreto na membrana celular [1,2].
Clínica
Na doença sistémica, diferentes órgãos são afectados, mas geralmente a doença pulmonar crónica leva ao aumento da morbilidade e à redução da esperança de vida. Devido à melhoria das possibilidades médicas, a esperança de vida tem aumentado continuamente nos últimos anos. A esperança média de vida na Europa é actualmente superior a 40 anos e continuará a aumentar, especialmente na geração detectada no rastreio de recém-nascidos na Suíça desde 2011. No entanto, o curso da doença é muito inconsistente, e os pacientes individuais ainda morrem de insuficiência pulmonar na idade adulta jovem. São comuns a todos os que sofrem de infecções bacterianas crónicas das vias respiratórias, que levam à destruição irreversível dos pulmões. Os pacientes sofrem de tosse, expectoração e uma limitação crescente da sua resiliência física. Além disso, é encontrada diarreia crónica com maldigestão devido a insuficiência pancreática exócrina, que afecta cerca de 85% dos doentes. Como consequência da desnutrição, mas também do aumento do trabalho de respiração, a incapacidade de obter resultados. Cerca de 10% já estão presentes com mecónio ileus após o nascimento, mas as pessoas mais velhas também podem sofrer de síndromes de obstrução intestinal recorrente. Apesar da terapia sintomática óptima, a cirrose hepática ou insuficiência pancreática endócrina com o desenvolvimento da diabetes mellitus também pode ocorrer no decurso posterior da doença. Os doentes masculinos com FC, em particular, são frequentemente afectados pela infertilidade [1,2].
Fisiopatologia
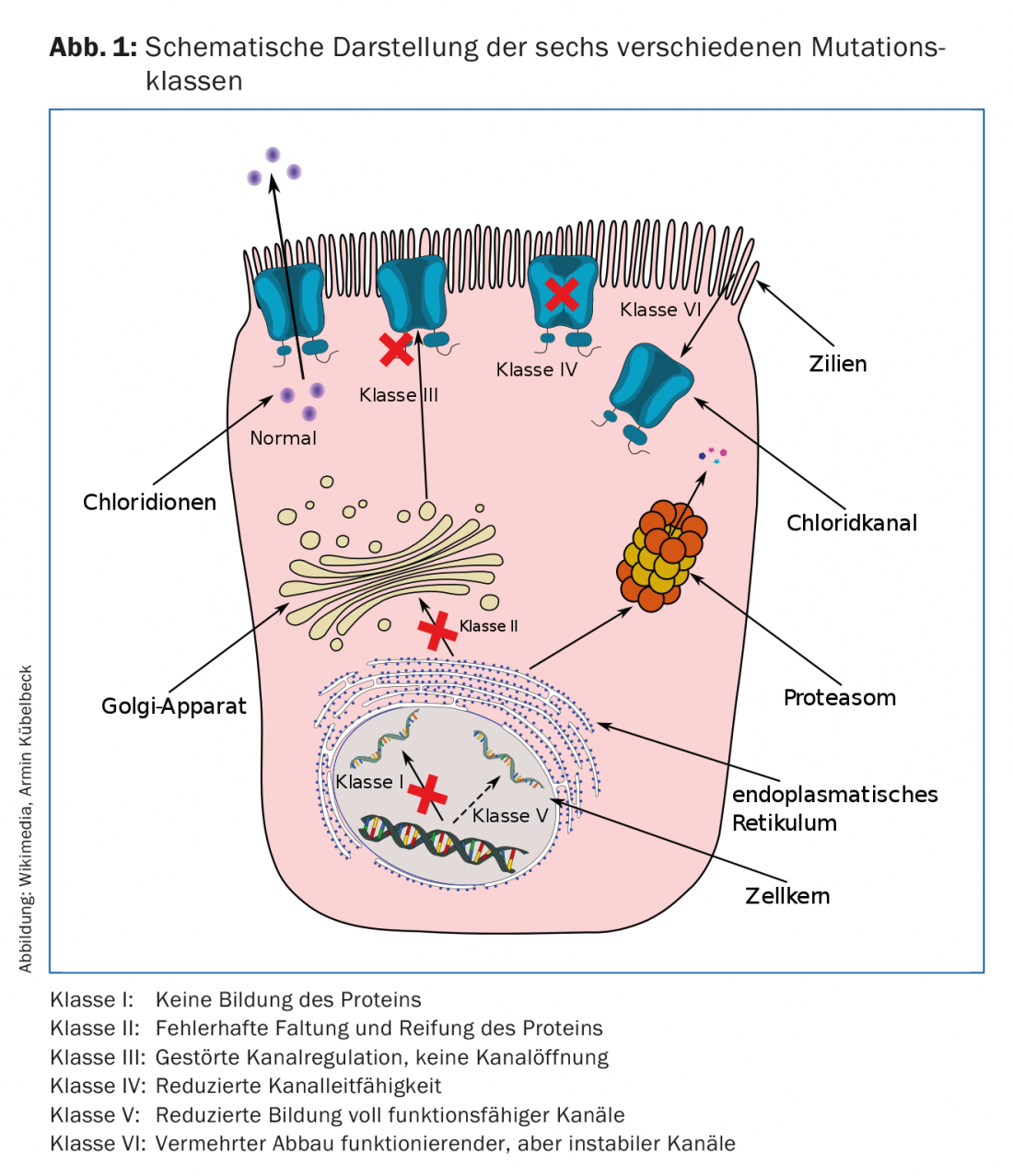
O gene CFTR nos códigos do cromossoma 7 para o canal do cloreto na membrana celular. As mutações genéticas, das quais mais de 2000 são agora conhecidas mas menos de 200 são claramente causadoras de doenças, causam um mau funcionamento ou ausência completa do canal de cloreto nas células epiteliais. As mutações no gene CFTR estão divididas em seis classes (Fig. 1), que diferem no seu patomecanismo. Algumas mutações, por exemplo, causam um fracasso quase completo da síntese da proteína CFTR. A mutação 3905insT, que ocorre em pessoas suíças ou de ascendência suíça (por exemplo, Amish na América do Norte), é atribuída a esta classe I e é a segunda mutação mais frequente na Suíça. Noutras mutações, a incorporação da proteína na membrana celular é impedida ou o canal iónico da proteína é bloqueado ou tem apenas uma condutividade limitada. A mutação mais frequente na Europa, mas também na Suíça (86% heterozigotos, 47% homozigotos) é atribuída à classe II e diz respeito a uma eliminação na posição 508 (F508del). As proteínas produzidas são dobradas incorrectamente aqui e degradadas antes de qualquer função ser assumida. A falha do canal do cloreto leva à interrupção do transporte transepitelial em todos os órgãos em que as células epiteliais exprimem o CFTR. Usando o epitélio das vias aéreas como exemplo, resulta uma cascata inflamatória. O aumento da viscosidade do muco causa uma depuração mucociliar deficiente, que por sua vez está associada a uma colonização bacteriana precoce e, no curso seguinte, crónica. A resposta imunitária do corpo causa recrutamento excessivo de granulócitos neutrófilos, libertação inadequada de elastase neutrófila e, por fim, destruição dos tecidos. Para além das duas mutações clássicas enumeradas como exemplos (F508del, 3905insT), que são responsáveis por um curso grave da doença, são agora também conhecidas mutações que estão associadas a um quadro clínico bastante suave – enquanto que no caso de outras mutações não é claro se são sequer causadoras de FC. A base de dados www.cftr2.org tenta fornecer uma visão geral continuamente actualizada das mutações causadoras de FC conhecidas e dos seus prováveis fenótipos [1,3,4].
Diagnósticos
Antes de 2011, cerca de 10% das crianças com FC foram diagnosticadas com mecónio ileus, mas a grande maioria só foi identificada durante o curso da doença quando apareceram sintomas de FC-suspiciosa. O padrão ouro para o diagnóstico é o teste do suor, ou seja, a determinação quantitativa da concentração de cloreto no suor após iontoforese de pilocarpina. Além disso, a medição da condutividade do suor é frequentemente utilizada como um teste de rastreio, uma vez que este teste funciona de forma rápida e fiável com uma quantidade significativamente menor de suor. Macroduto® e Nanoduto® são sistemas de teste disponíveis comercialmente. Para efeitos de garantia de qualidade, a realização de testes de soldadura deve ser reservada a centros apropriados.
Em 2011, a CF foi incluída no programa de rastreio do recém-nascido suíço. O tripsinogénio imunoactivo (IRT) é medido no sangue do calcanhar. Se o limite for ultrapassado, os recém-nascidos são encaminhados para um centro CF pediátrico. Aí, o diagnóstico é confirmado ou excluído com testes de suor e, se necessário, outros exames (determinação da elastase pancreática, análise genética).
Até 2016, 143 crianças tinham sido diagnosticadas com FC através de rastreio de recém-nascidos. Num total estimado de 900 doentes com FC, cerca de 15% de todos os doentes já puderam ser detectados com a ajuda do rastreio de recém-nascidos. Este valor continuará a aumentar de forma constante.
Para o GP em particular, contudo, é importante notar que, devido aos diferentes fenótipos, podem existir casos diagnosticados na infância, bem como aqueles com um curso ligeiro ou com CFTR residual mensurável. Os indivíduos só podem, portanto, tornar-se aparentes na idade adulta com sintomas atípicos tais como pancreatite, sinusite, pólipos nasais, bronquiectasias difusas e/ou um desejo não satisfeito de ter filhos. Por conseguinte, o médico de clínica geral deve também considerar a FC como um possível diagnóstico diferencial em caso de sintomas correspondentes [1,5–7].
Terapia e cuidados
De acordo com directrizes internacionais, os pacientes devem estar ligados a centros especializados. Os pacientes são vistos a intervalos de 3 meses. Dependendo do curso individual da doença, a terapia deve ser revista e ajustada. Para além da história médica e do exame clínico, as medições da função pulmonar (dependendo da infra-estrutura, pletismografia corporal, espirometria, lavagem de respiração múltipla N2) e os testes microbiológicos (expectoração ou esfregaço de garganta) fazem parte dos exames de rotina, que são complementados a intervalos maiores por imagens dos pulmões e do abdómen, análises ao sangue, cargas de glicose e medições da densidade óssea. O objectivo é detectar alterações numa fase precoce para que possam ser contrariadas e para que se possa evitar uma maior deterioração.
O tratamento básico é complexo e inclui a desobstrução intensiva das vias aéreas por inalação e fisioterapia respiratória especial (solução salina hipertónica, rhDNAse se necessário, “técnica especial de desobstrução das vias aéreas”), substituição de enzimas pancreáticas (lipase) e terapia nutricional (substituição de vitaminas lipossolúveis, dieta rica em calorias), bem como tratamento antimicrobiano agressivo (inalação e/ou aplicação sistémica).
Dependendo da idade do paciente e das circunstâncias que o acompanham, é também necessário um aconselhamento contínuo e direccionado para poder aceitar da melhor forma os desafios da doença e os seus efeitos sobre o modo de vida. Para além da doença óbvia e do conteúdo relacionado com a terapia, os pacientes (e dependendo da sua idade, também os seus tutores legais) também necessitam de apoio no tratamento de questões sociais, psicológicas, financeiras e relacionadas com seguros (invalidez e/ou seguro de saúde), na organização de cuidados infantis, frequência de jardins de infância e escolas, formação e trabalho, viagens, o desejo dos pais de ter filhos novamente ou o planeamento familiar do próprio paciente adulto, etc.
O facto de as pessoas afectadas estarem, felizmente, a ficar cada vez mais velhas deve também ser tido em conta. De repente, os pacientes, mas também as equipas de tratamento, são expostos a problemas internos que surgem independentemente da FC, mas influenciam o seu curso. Para responder a estas exigências, é necessária uma abordagem terapêutica multidisciplinar, que só pode ser fornecida por um centro CF [8,9].
Perspectivas
Em 2014, o Swissmedic aprovou o medicamento ivacaftor (Kalydeco®) para pacientes com a mutação de classe III G551D. Pela primeira vez, uma terapia causal com um aumento significativo da função do canal do cloro, mensurável numa melhoria acentuada no teste de suor e na função pulmonar, poderia ser utilizada nos poucos pacientes com esta mutação (aproximadamente 4-5% de todos os pacientes em todo o mundo). Outros moduladores CFTR estão actualmente a ser testados. Resta a esperança de que uma terapia causal específica da mutação esteja disponível para um grupo maior de pacientes no futuro [10].
Mensagens Take-Home
- Devido a melhorias na terapia sintomática e na gestão de doenças, a qualidade de vida e a esperança de vida estão continuamente a aumentar.
- Já hoje, metade de todos os pacientes da Swiss CF estão na idade adulta.
- O rastreio do recém-nascido para a FC introduzido em 2011 permite o diagnóstico precoce. Isto significa que a terapia básica pode ser iniciada antes de ocorrerem as primeiras alterações relacionadas com a doença.
- São possíveis cursos oligossintomáticos e atípicos. Por conseguinte, a FC deve também ser considerada como um possível diagnóstico diferencial na idade adulta.
- A clarificação e os cuidados adicionais devem sempre ter lugar em cooperação com um centro CF.
Literatura:
- Elborn JS: Fibrose cística. Lancet 2016 19 de Novembro; 388(10059): 2519-2531.
- Zolin A, et al: Relatório Anual do ECFSPR 2014. 2016.
- Hergersberg M, et al.: Uma nova mutação, 3905insT, é responsável por 4,8% dos 1173 cromossomas CF na Suíça e causa um fenótipo grave. Hum Genet 1997 Ago; 100(2): 220-223.
- CFTR2. www.cftr2.org
- Torresani T, et al: Rastreio do recém-nascido para fibrose cística na Suiça após análise de um estudo piloto de 4 meses. J Cyst Fibros 2013; 12(6): 667-674.
- Rastreio de recém-nascidos na Suíça. www.neoscreening.ch
- Barben J, et al: Rastreio de recém-nascidos para fibrose cística – uma história de sucesso. Schweiz Med Forum 2013; 13(49): 1010-1012.
- Smyth AR, et al: European Cystic Fibrosis Society Standards of Care: Best Practice guidelines. J Cyst Fibros 2014 Maio; 13(Suppl 1): S23-42.
- Elborn JS, et al: Relatório da task force da Sociedade Respiratória Europeia/Sociedade Europeia de Fibrose Cística sobre o cuidado de adultos com fibrose cística. Eur Respir J 2016 Fev; 47(2): 420-428.
- Ramsey BW, et al: Um potenciador CFTR em pacientes com fibrose cística e a mutação G551D. N Engl J Med 2011 Nov 3; 365(18): 1663-1672.
PRÁTICA DO GP 2017; 12(11): 31-33









