
Una linfoadenopatia persistente è una chiara indicazione per un’ulteriore diagnosi istopatologica. Un’anamnesi medica dettagliata, nonché la conoscenza delle malattie precedenti, delle manipolazioni e delle terapie farmacologiche sono essenziali per una diagnosi definitiva di linfoma. Ci sono casi che si trovano in aree grigie in termini di classificazione. La presenza di riarrangiamenti MYC è un biomarcatore prognostico indiscusso nel DLBCL, così come la coespressione fenotipica delle proteine myc e bcl2. La risposta alla terapia con rituximab è legata all’espressione del CD20. Gli studi molecolari stanno producendo nuovi parametri genetici predittivi per la risposta specifica alle terapie mirate nel linfoma.
Con una quota relativa del 5%, i linfomi rappresentano il quinto gruppo più comune di malattie maligne in entrambi i sessi. Soprattutto i linfomi a cellule B mature (in passato “linfomi non-Hodgkin”) mostrano l’incidenza di malignità in maggiore crescita nel mondo industrializzato, dopo il melanoma.
Le ragioni sono sconosciute, ma potrebbero essere legate all’aumento dell’aspettativa di vita, alla crescente incidenza di malattie autoimmuni e all’uso diffuso di (nuovi) immunosoppressori, o all’aumento dell’esposizione a determinati pesticidi ed erbicidi. L’incidenza del linfoma è attualmente di circa 25 casi/100.000 abitanti/anno.
I progressi della terapia oncologica sono evidenti nel caso dei linfomi. I tassi di sopravvivenza a 5 anni specifici per la malattia per i linfomi nodali più comuni, come il linfoma diffuso a grandi cellule B (DLBCL), il linfoma follicolare (FL), il linfoma classico di Hodgkin (HL) e il linfoma linfoblastico a cellule B (B-LBL, equivalente nodale della leucemia linfoblastica acuta a cellule B), sono di circa il 60% (DLBCL) e di circa l’80% (le altre entità). Questo spiega l’alta prevalenza del linfoma.
Definizione
I linfomi sono definiti come malattie maligne, neoplastiche dei linfociti B, T o NK immaturi o maturi negli organi del sistema linfatico (nodali) o al di fuori di tali organi (extranodali). Possono essere leucemici o senza essudazione (linfomi in senso stretto).
Clinica
I linfomi nodali si presentano con una linfadenopatia localizzata o generalizzata, persistente (più di tre settimane), spesso progressiva, con o senza sintomi generali (B) (febbre, sudorazione notturna, perdita di peso), coinvolgimento degli organi, alterazioni cutanee (prurito, eritema) o segni di insufficienza del midollo osseo (anemia, petecchie, tendenza alle infezioni).
Una linfoadenopatia persistente, soprattutto se accompagnata dai sintomi citati, è quindi una chiara indicazione per un’ulteriore diagnosi.
Diagnostica
La diagnostica basata sui tessuti è indispensabile nei linfomi, in quanto le alterazioni istopatologiche dei tessuti sono le pietre miliari della determinazione della dignità e dell’entità. In base a questi cambiamenti, è possibile distinguere tra benigni e maligni e il grado di maturazione dei linfociti colpiti dalla microscopia ottica convenzionale. Mediante ulteriori metodi microscopici in situ, come l’immunoistochimica (espressione di proteine) o l’ibridazione in situ a fluorescenza (aberrazioni cromosomiche ricorrenti), è possibile determinare l’appartenenza al lignaggio (lignaggio B, T o NK), l’esatto stadio di sviluppo (ad esempio, cellule B del centro germinale) e le espressioni dei marcatori patologici (ad esempio, l’espressione dei marcatori delle cellule T sulle cellule B, come nella leucemia linfocitica cronica a cellule B). [B-CLL]) o traslocazione cromosomica esistente (ad esempio, t[14;18] alla FL) può essere determinata e si può fare una diagnosi esatta. Questo è un prerequisito fondamentale per una terapia oncologica specifica. Nei casi diagnosticamente difficili, si può ottenere il DNA dal materiale (fissato in formalina e incluso in paraffina) e analizzare ulteriormente la clonalità delle cellule B e T, le traslocazioni e le mutazioni puntiformi.
Classificazione
I linfomi sono classificati secondo l’attuale classificazione dell’OMS. Il principio fondamentale è la diagnosi integrativa, che considera l’inclusione della morfologia istopatologica del linfoma, i fenotipi (modello di espressione proteica), i genotipi (aberrazioni cromosomiche ricorrenti) e la clinica di pari importanza nella classificazione delle entità.
La separazione primaria dei linfomi in linfomi Hodgkin e non Hodgkin è stata abbandonata. A causa della sua morfologia caratteristica, della presentazione clinica e dell’eccellente risposta a terapie specifiche, l’HL continua ad essere gestito come un’entità distinta.
Nuove categorie: A causa della complessità biologica di alcune malattie, l’OMS ha introdotto due cosiddette “categorie di zona grigia”:
- Linfomi a cellule B non classificabili con caratteristiche intermedie tra un DLBCL e un HL
- Linfomi a cellule B non classificabili con caratteristiche intermedie tra un DLBCL e un linfoma di Burkitt (BL).
Oltre alla sovrapposizione clinica e morfologica tra le singole entità di entrambe le categorie, gli studi molecolari mostrano una significativa sovrapposizione di geni espressi tra i linfomi primari mediastinici a grandi cellule B e HL, da un lato, e tra i singoli DLBCL e BL, dall’altro.
Il decorso clinico sfavorevole dei casi di DLBCL altamente proliferativi con disposizione del gene MYC (Fig. 1a) giustifica ulteriormente l’introduzione di questa categoria di zona grigia per seguire questi casi, che hanno una risposta inadeguata alle terapie DLBCL standard, in studi terapeutici prospettici.
Categorie diagnostiche basate su parametri clinici: Nella classificazione dell’OMS, sono stati presi in forte considerazione i contesti legati al paziente, come l’età, la terapia farmacologica precedente o in corso, soprattutto quella immunosoppressiva, e la localizzazione. Quattro entità sono definite dall’età dei pazienti:
- FL pediatrico
- Linfoma a cellule B della zona marginale nodale (MZL) pediatrico
- Malattia linfoproliferativa a cellule T dell’infanzia associata al virus di Epstein-Barr (EBV)
- DLBCL associato all’EBV negli anziani.
Il razionale per l’introduzione di queste entità è l’associazione della comparsa con l’immaturità o la senescenza del sistema immunitario. Mentre i primi tre elencati sono decisamente rari, la percentuale relativa di questi ultimi è del 3% di tutti i DLBCL. Sebbene gli studi dell’Estremo Oriente indichino una chiara aggressività di questo linfoma, i nostri dati provenienti dall’Europa centrale mostrano una particolare aggressività clinica solo in casi individuali.
Riflettendo la frequenza e l’eterogeneità del DLBCL, nella nuova classificazione sono state definite ulteriori varianti clinicopatologiche. Per alcune di queste varianti, come il DLBCL associato a infiammazione cronica (pitorace, osteomielite cronica, impianti infetti di corpi estranei, ad esempio protesi vascolari/articolari, ulcere cutanee croniche, articolazioni artritiche nell’artrite reumatoide), la conoscenza della clinica è indispensabile in termini di classificazione. (Fig. 1b). Poiché il 70% di questi linfomi è associato all’EBV e si verifica soprattutto nei pazienti anziani, la differenziazione dal DLBCL associato all’EBV negli anziani è possibile solo sulla base di prove anamnestiche.
La conoscenza della clinica è indispensabile anche per la diagnosi delle malattie linfoproliferative iatrogene, associate all’immunodeficienza (terapia con immunosoppressori), nonché per la diagnosi delle linfoproliferazioni post-trapianto (trapianto di organi o di midollo osseo allogenico). Lo stesso vale per la diagnosi dei “linfomi a piccole cellule B” secondari, trasformati (B-CLL, MZL, FL), che possono trasformarsi in un DLBCL, in un linfoma a cellule B non classificabile con caratteristiche intermedie tra un DLBCL e un BL, nonché in un HL. L’indicazione anamnestica di un precedente linfoma indolente del gruppo dei “linfomi a piccole cellule B” è decisiva per la corretta classificazione di tali lesioni. Questo è significativo perché i DLBCL o HL (Fig. 1c), che si trasformano da questi linfomi a cellule B, hanno un decorso significativamente più aggressivo rispetto ai loro analoghi de novo.
Prognosi e previsione
Nell’ultimo decennio, numerosi studi hanno cercato di stabilire fattori prognostici significativi legati al tumore nei linfomi nodali più comuni, come DLBCL, FL e HL. Tuttavia, i fattori prognostici clinici noti, come l’International Prognostic Index (IPI) nel DLBCL, il FL International Prognostic Index (FLIPI) e l’International Prognostic Score (IPS) nel HL, non sono stati superati dall’inclusione di fattori prognostici legati al tumore. Ad eccezione di alcune varianti di DLBCL che sono associate a una prognosi piuttosto peggiore, come il DLBCL ricco di cellule T e istociti, la granulomatosi linfomatoide e il DLBCL intravascolare, solo il rilevamento dei riarrangiamenti MYC è un parametro prognostico indiscusso legato al tumore nel DLBCL. Tre studi indipendenti, nuovi e di grandi dimensioni, hanno dimostrato il ruolo prognostico sfavorevole della coespressione fenotipica delle proteine myc e bcl2 (il cosiddetto “DLBCL fenotipico double-hit”). Gli studi su HL e FL hanno mostrato un effetto prognostico della composizione delle cellule T di base. Recenti studi di espressione genica hanno anche dimostrato che le firme dei macrofagi associati al tumore possono influenzare significativamente la sopravvivenza nell’HL, come si evince morfologicamente dagli alti livelli di macrofagi tissutali nei pazienti con prognosi sfavorevole (Fig. 1d).
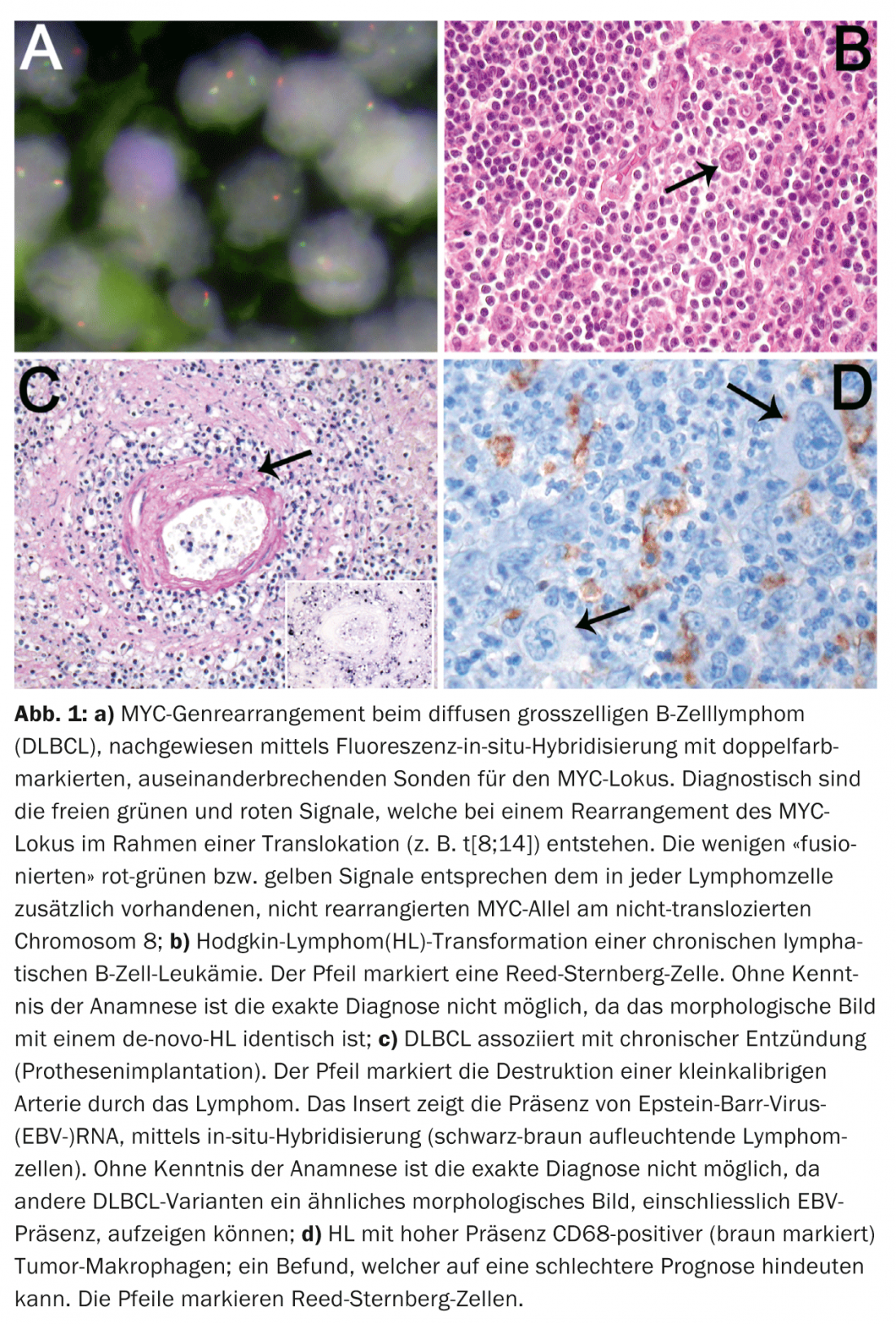
In sintesi, i marcatori prognostici nel linfoma non sono ancora pronti per la pratica quotidiana. Tuttavia, la ricerca di parametri prognostici associati al microambiente tumorale sembra promettente, soprattutto perché questo ambiente potrebbe essere manipolato terapeuticamente senza temere che il tumore sviluppi resistenza.
Un altro campo di ricerca riguarda la creazione di marcatori predittivi, ossia biomarcatori che indicano la risposta o la non risposta a una terapia. Sebbene gli studi clinici non abbiano studiato in modo specifico l’espressione del CD20 nell’istituzione dell’anticorpo terapeutico anti-CD20 rituximab, l’esperienza dimostra che solo i linfomi a cellule B che esprimono il CD20 rispondono a questa terapia. La determinazione dell’espressione del CD20 nei linfomi è quindi un esempio di marcatore predittivo determinabile dal punto di vista istopatologico.
Dati recenti suggeriscono che la sensibilità specifica agli inibitori della chinasi di Bruton (ad esempio ibrutinib), agli agenti che promuovono l’apoptosi (ad esempio obatoclax) e agli inibitori della fosfoinositide 3-chinasi (compresi gli inibitori di mTOR, come everolimus) può essere prevista da cambiamenti genetici specifici nelle cellule linfoidi.
Prof. Alexandar Tzankov, MD
Prof. Dr. med. Stephan Dirnhofer
Letteratura:
- Roman E, Smith AG: Istopatologia 2011; 58: 4-14.
- Swerdlow SH, et al: Classificazione OMS dei tumori dei tessuti ematopoietici e linfoidi. Lione: IARC; 2008.
- Hoeller S, et al: Hum Pathol 2010; 41: 352-357.
- Tzankov A, et al.: Mod Pathol 2013; doi: 10.1038/modpathol.2013
- Hu S, et al: Blood 2013; 121: 4021-4031.
- Steidl C, et al: N Engl J Med 2010; 362: 875-885.
- Tzankov A, et al: Haematologica 2008; 93: 193-200.
- Rahal R, et al: Nat Med 2014; 20: 87-92.
- Wenzel SS, et al: Leukemia 2013; 27: 1381-1390.
- Pfeifer M, et al: Proc Natl Acad Sci U S A. 2013; 110: 12420-12425.
InFo Oncologia & Ematologia 2014; 2(2): 5-7









