
La diagnosi precoce, la stratificazione del rischio e la terapia su misura per la sclerosi sistemica (SSc) sono di grande importanza per poter contrastare tempestivamente le limitazioni legate alla malattia e le manifestazioni potenzialmente letali. La malattia polmonare interstiziale (ILD) è una manifestazione d’organo della SSc con una mortalità significativa. Oltre agli immunosoppressori, il farmaco antifibrotico nintedanib è disponibile da tempo in Svizzera e tocilizumab è stato recentemente approvato negli Stati Uniti sulla base di risultati positivi di fase III.
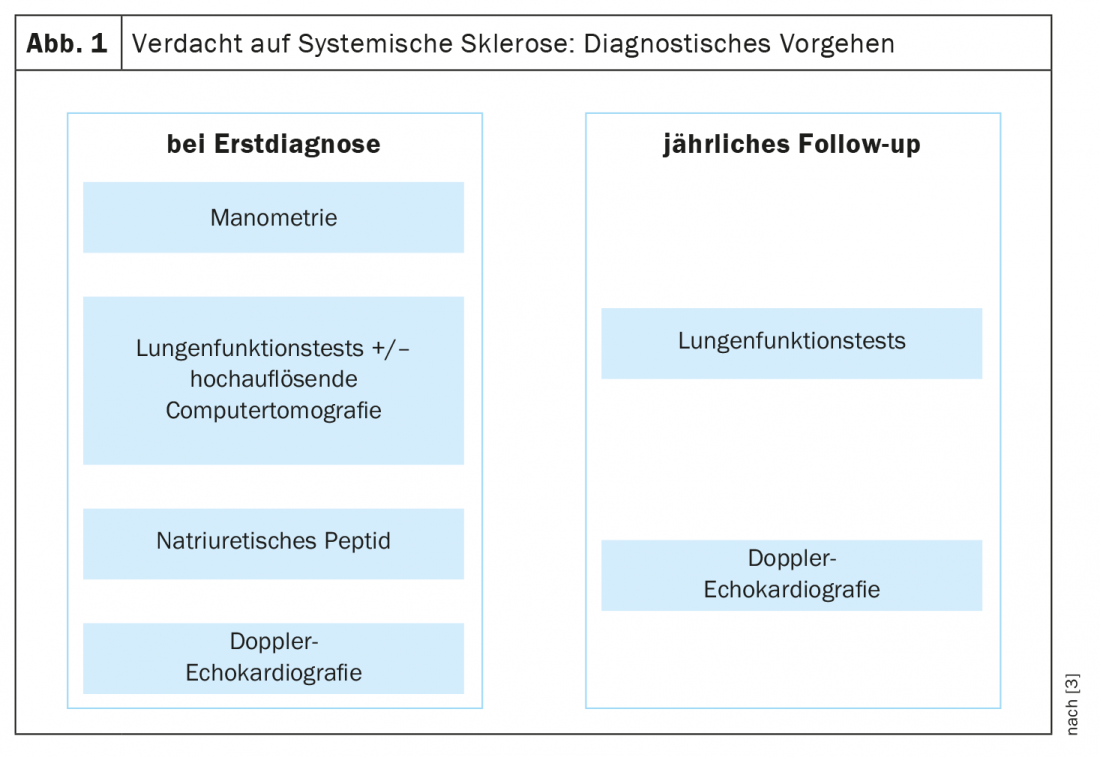
Per la diagnosi precoce della sclerosi sistemica, alcuni anni fa sono stati stabiliti i criteri VEDOSS (“Very Early Diagnosis of Systemic Sclerosis”) [1,2]. La sindrome di Raynaud, il gonfiore delle dita (“dita gonfie”), i cambiamenti nei capillari della piega ungueale e il rilevamento degli autoanticorpi antinucleari (ANA) sono predittivi della sclerosi sistemica (SSc), ha spiegato la Dr.ssa Hanna Grasshoff, Clinica di Reumatologia e Immunologia Clinica, Ospedale Universitario Schleswig-Holstein, Lubecca [1]. I criteri di classificazione stabiliti nel 2013 dall’American College of Rheumatology (ACR) e dalla European League Against Rheumatism (EULAR) sono ancora validi [1,16]. Gli ANA più comuni associati alla SSc includono gli anti-centromero-Ak (ACA) e gli anti-topoisomerasi-Ak (ATA, Scl70). Gli attuali risultati empirici confermano che i criteri VEDOSS sono adatti per la stratificazione del rischio dei pazienti. Questo è stato dimostrato anche da uno studio pubblicato nel 2021 sull’European Journal of Internal Medicine [3]. Il relatore riassume la procedura diagnostica come segue (Fig. 1): “I pazienti vengono sottoposti a screening con manometria esofagea, test di funzionalità polmonare e, se necessario, tomografia computerizzata ad alta risoluzione in caso di anomalie, nonché peptide natriuretico ed ecocardiografia Doppler” [1]. Può essere utile un ECG supplementare [1]. Durante il corso si raccomandano test annuali di funzionalità polmonare ed ecocardiografie Doppler. Per quanto riguarda l’ipertensione arteriosa polmonare, compresa la SSc-PAH, è stata pubblicata una nuova linea guida ESC/ERS nel 2022 [17]. I valori target precedenti sono stati modificati e sono ora i seguenti: mPAP (pressione arteriosa polmonare media) >20 mmHg, PAWP (pressione di cuneo arterioso polmonare) ≤15 mmHg, PVR (resistenza vascolare polmonare) ≥ 2 WU (unità Wood*).
* unità di misura tradizionale delle resistenze dei vasi.
“Utilizzare le “Finestre di opportunità
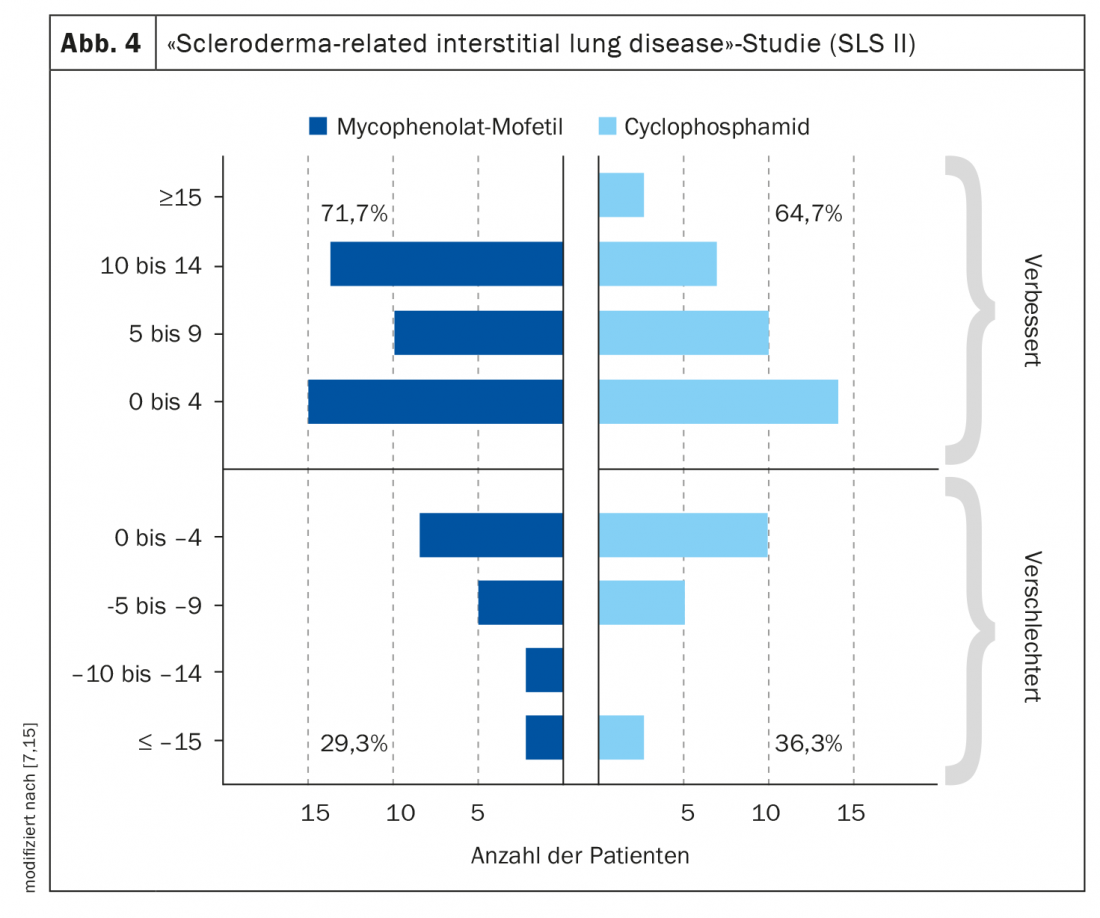
La distinzione tra SSc cutanea limitata e SSc cutanea diffusa è ancora rilevante. Secondo le raccomandazioni dell’EULAR, la procedura terapeutica dipende dal coinvolgimento dell’organo [5]: 1. Sindrome di Raynaud, 2. ulcere digitali, 3. SSc-PAH, 4. manifestazioni cutanee e polmonari, 5. crisi renale, 6. manifestazione gastrointestinale. I due farmaci più comunemente utilizzati sono la ciclofosfamide e il micofenolato mofetile (MMF), sulla base di due studi randomizzati controllati con risultati di efficacia simili [6,7]. Nello Scleroderma Lung Study (SLS) II, la ciclofosfamide e l’MMF hanno ottenuto un’efficacia paragonabile, ma l’MMF aveva un profilo di sicurezza e tollerabilità a lungo termine migliore (Fig. 4). Pertanto, l’MMF viene utilizzato più frequentemente nella pratica clinica. Di grande interesse, naturalmente, è la questione delle implicazioni di una diagnosi precoce per la terapia. In uno studio di coorte retrospettivo pubblicato nel 2022, i pazienti affetti da SSc che hanno manifestato una SSc cutanea diffusa o una malattia polmonare interstiziale entro 6 anni dall’esordio della malattia sono stati suddivisi in gruppi di intervento precoce e ritardato in base alla durata della malattia. Nel primo caso, il trattamento è stato iniziato entro ≤18 mesi dall’esordio della malattia, nel secondo solo >18 mesi dopo [4]. Le opzioni terapeutiche utilizzate sono state ciclofosfamide, MMF, metotrexato o tocilizumab. Nel gruppo di intervento precoce, la malattia attiva è diminuita significativamente dal 79% al 42% (p=0,007), mentre il cambiamento nel gruppo di intervento ritardato non è stato statisticamente significativo (dal 68% al 42%; p=0,11). Nel complesso, i risultati di questo studio supportano l’ipotesi che esista una “finestra di opportunità” per le opzioni terapeutiche nei pazienti con SSc.
SSc-ILD – spettro terapeutico esteso per una complicanza potenzialmente letale
Per le manifestazioni cutanee e polmonari, l’EULAR raccomanda le seguenti opzioni farmacologiche: Metotrexato, ciclofosfamide, trapianto autologo di cellule staminali ematopoietiche, eventualmente MMF, eventualmente azatioprina. La malattia polmonare interstiziale (ILD) nella sclerosi sistemica (SSc-ILD) è attualmente la più comune causa di morte associata alla malattia nei pazienti con SSc [8]. La prevalenza di ILD come complicanza della SSc è di circa il 50% in una recente coorte basata sulla popolazione [9]. La principale terapia farmacologica utilizzata finora è stata quella degli immunosoppressori, con l’MMF che è il farmaco più utilizzato a livello internazionale [10,11]. Studi recenti suggeriscono che nintedanib e tocilizumab possono contribuire a rallentare il deterioramento della funzione polmonare nella SSc-ILD [12]. Nintedanib è un inibitore della tirosin-chinasi antifibrotico, utilizzato nella malattia polmonare interstiziale per prevenire la cicatrizzazione del tessuto polmonare. In Svizzera, nintedanib (Ofev®) è stato approvato dal 2020 per il trattamento della malattia polmonare interstiziale associata alla sclerosi sistemica [12]. Il dosaggio consigliato è di 150 mg due volte al giorno, a intervalli di circa 12 ore. Tocilizumab è stato approvato dalla FDA statunitense per il trattamento della SSc-ILD sulla base dei dati di uno studio di fase III [13].
La medicina di precisione indica la strada per il futuro
Un documento di posizione sul trapianto autologo di cellule staminali ematopoietiche (ahSCT) per la sclerosi sistemica è stato pubblicato dal Gruppo di lavoro sulla terapia con cellule staminali della Società tedesca di reumatologia [14]. Di conseguenza, la ahSZT è ragionevole alle seguenti condizioni: durata massima della malattia di 4 anni, mRSS di min. 15, coinvolgimento di organi interni o altri fattori prognosticamente sfavorevoli, risposta insufficiente alla ciclofosfamide o al MMF. La SSc è una malattia molto eterogenea. “Abbiamo bisogno di una stratificazione molecolare a diversi livelli OMICS nel lungo termine”, afferma il dottor Grasshoff [1]. È qui che entra in gioco la medicina di precisione. “L’obiettivo sarebbe quello di trattare la vasculopatia in modo vasoattivo, l’infiammazione in modo immunomodulatorio e la fibrosi in modo antifibrotico”, ha detto [1]. Gli studi che correlano la firma genica intrinseca con la risposta a diversi farmaci sono un approccio promettente per una strategia di trattamento mirata a un sottogruppo specifico con un profilo beneficio-rischio ottimizzato.
Congresso: Congresso tedesco di reumatologia
Letteratura:
- “La sclerosi sistemica – una malattia eterogenea”, Dr. Hanna Grasshoff, Congresso tedesco di reumatologia, 31.08.-03.09.2022.
- Minier T, et al: Ann Rheum Dis 2014; 73(12): 2087-2093.
- Gonzalez Garcia A, Callejas-Rubio JL: European Journal of Internal Medicine 97(Suppl 113); DOI:10.1016/j.ejim.2021.12.012
- Yomono K, Kuwana M: Rheumatology (Oxford) 2022; 61(9): 3677-3685.
- Kowal-Bielecka O, et al: Ann Rheum Dis 2017; 76(8): 1327-1339.
- Tashkin DP, et al: NEJM 2006 (354): 2655-2666.
- Tashkin DP, et al: Lancet Respir Med 2016 (4): 708-719.
- Elhai M, et al: Ann Rheum Dis 2019; 78: 979-987.
- Hoffmann-Vold AM, et al: Am J Respir Crit Care Med 2019; 200: 1258-1266.
- Fernández-Codina A, et al: Arthritis Rheumatol 2018; 70: 1820-1828.
- Khanna D, et al: [abstract]. Arthritis Rheumatol 2018, 70 (suppl 9). https://acrabstracts.org/
- Informazioni sui farmaci, www.swissmedicinfo.ch, (ultimo accesso 07.11.2022)
- Khanna D, et al: Lancet Respir Med 2020; 8: 963-974
- Alexander T, Burmester G: Giornale di Reumatologia. Edizione 5/2020.
- Schneider U, et al: Z Rheumatol 2021; 80: 868-878.
- van den Hoogen F, et al: Arthritis Rheum 2013; 65(11): 2737-2347.
- Humbert M, et al: Gruppo di documenti scientifici ESC/ERS. EHJ 2022; 43(38): 3618-3731.
PRATICA GP 2022; 17(11): 16-17









