A taquicardia ventricular polimórfica catecolaminérgica (CPVT) é uma doença rara dos canais iónicos sem cardiopatia estrutural detectável macroscopicamente. Ocorre exclusivamente na infância e adolescência e tem uma elevada taxa de mortalidade se não for tratada. Por conseguinte, a terapia do coração estruturalmente inconspícuo é de particular importância.
A taquicardia ventricular polimórfica catecolaminérgica (CPVT) é uma doença rara do canal iónico sem cardiopatia estrutural detectável macroscopicamente, com uma prevalência de 1:5000-10.000. Descrita pela primeira vez em 1975 [1], não foi caracterizada como uma entidade separada até 1995 [2]. Característica é a ocorrência de extra-sístoles ventriculares polimórficas e taquicardia ventricular (VT), bem como VT bidireccional sob stress físico ou mental com um ECG em repouso sem precedentes. Embora existam relatos de casos individuais de primeiras manifestações sintomáticas até à idade de 40 anos, trata-se predominantemente de uma doença de crianças e adolescentes. Dependendo do subtipo genético, a primeira manifestação ocorre entre os 2 e os 20 anos de idade, com um agrupamento familiar em 30% dos casos. Homens e mulheres são igualmente afectados, com uma apresentação anterior em homens [3]. O rastreio familiar deve ser sempre feito [4].
Fisiopatologia
Embora já tenha havido grandes progressos na caracterização dos mecanismos de doença subjacentes desde a descrição inicial, estes ainda não são totalmente compreendidos.
Defeitos em cálcio diastólico (Ca2+) libertados do retículo sarcoplasmático (SR) resultam em sobrecarga de Ca2+ na célula miocárdica. Em indivíduos saudáveis, o fluxo extracelular de Ca2+ através de canais do tipo L, desencadeado pelo potencial de acção, resulta na libertação de Ca2+ acionada por Ca2+ a partir do SR através da ativação do receptor ryanodine (RYR2). Isto leva à contracção do miocárdio e à fase de planalto do potencial de acção com subsequente transporte de Ca2+ de volta para o espaço extracelular e para o SR. No CPVT, contudo, um defeito autossómico dominante do RYR2, entre outros, leva a uma libertação espontânea diastólica Ca2+ do SR. Isto leva a uma inversão do transporte de Ca2+ para a célula e a pós-depolarizações tardias, que constituem a base do CPVT. Se a estimulaçãoadrenérgica β é também desencadeada pelo stress, desenvolvem-se as extra-sístoles ou taquicardias polimórficas ventriculares características [5].
Além das mutações no gene RYR2, que são detectáveis em cerca de 50-55% dos pacientes com CPVT, existem outras mutações da calsequestrina cardíaca (CASQ2) e da triadina (TRDN), dois outros componentes do orçamento celular Ca2+ [5]. Além disso, a disfunção dos canais de potássio (KCNJ2) também pode levar ao CPVT. Dependendo do defeito genético detectado, é possível distinguir entre vários subtipos. Globalmente, no entanto, os defeitos genéticos responsáveis só podem ser detectados em cerca de 60% das pessoas afectadas [5].
Diagnóstico
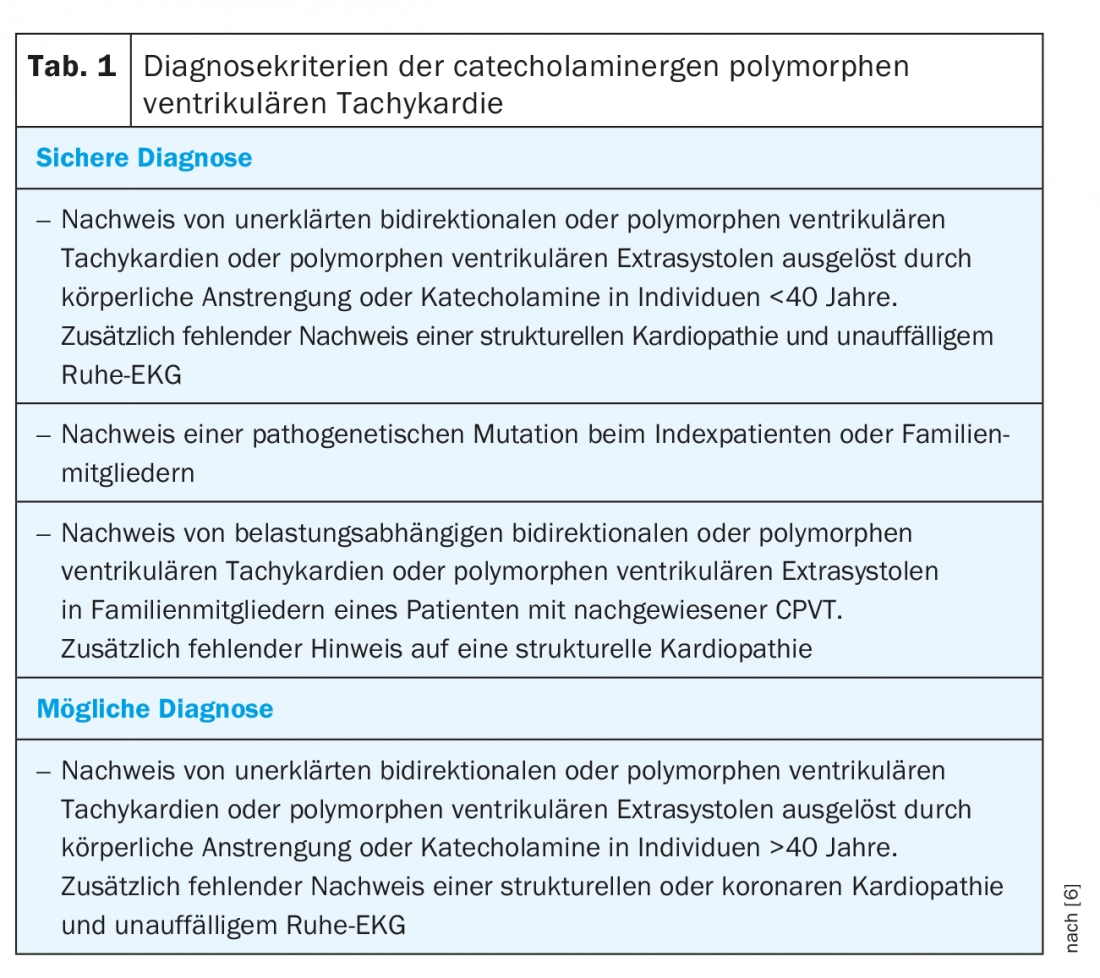
Os critérios de diagnóstico das sociedades profissionais estão resumidos no quadro 1 [6]. No entanto, é muito mais difícil do que aplicar os critérios de diagnóstico para identificar possíveis sofredores. As apresentações clínicas típicas são crianças entre os 2-20 anos de idade com síncope experiente ou que sobreviveram à morte cardíaca súbita (SCD) durante o stress físico ou emocional [2]. Como a síncope relacionada com o CPVT também pode causar convulsões e incontinência, as crianças são muitas vezes tratadas erroneamente por epilepsia. O CPVT é então diagnosticado tardiamente na ausência de eficácia dos medicamentos antiepilépticos. Do mesmo modo, um agrupamento familiar de síncope de exercício ou SCD e epilepsia familiar refratária deve sugerir um possível CPVT familiar. A apresentação atípica no contexto do rastreio cardiovascular por ergometria é possível na idade adulta, mas muito menos frequente.
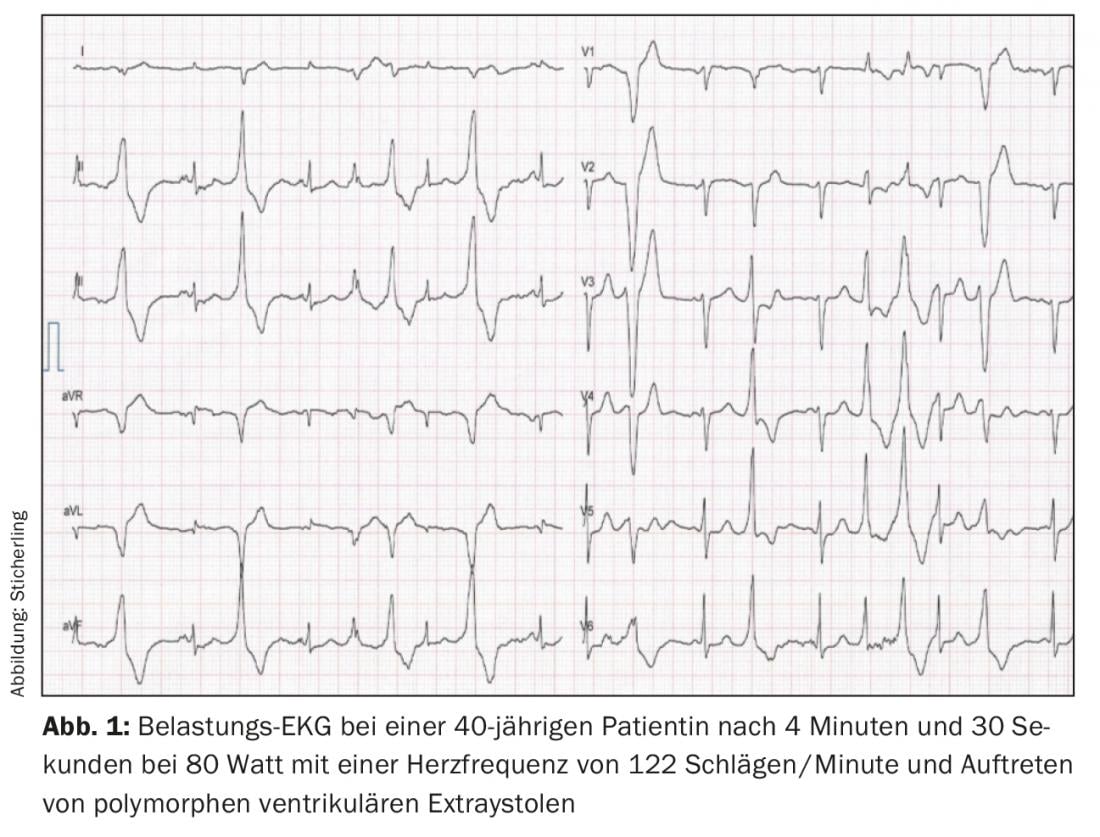
O padrão de ouro para o diagnóstico é a ergometria de exercício. Tipicamente, pode ser determinado um limiar de frequência cardíaca reproduzível individualmente entre 110-130 batimentos/minuto, acima do qual ocorrem as primeiras extra-sístoles ventriculares isoladas com um intervalo de acoplamento de aproximadamente 400 ms. As extra-sístoles mostram predominantemente um eixo superior com bloco de ramo esquerdo ou um eixo inferior com bloco de ramo direito [4,7]. Com o aumento do stress e do ritmo cardíaco, ocorrem extrassístoles monomórficas mais frequentes, seguidas de bigeminy e finalmente de extrassístoles polimórficas e/ou VT não-sustentadas (Fig. 1). Raramente pode ocorrer VT polimórfico sustentado ou fibrilação ventricular. O teste de stress deve portanto ser interrompido se os sintomas arritmogénicos aumentarem ou se a duração do VT não-sustentado aumentar [7,8]. Um ECG ambulatorial de longo prazo pode ter valor diagnóstico adicional em pacientes muito jovens, em pacientes que não podem realizar um ECG de exercício, ou em pacientes com um ECG de exercício negativo, mas ainda suspeitos de CPVT. No entanto, a sensibilidade é menor do que com o ECG de stress [9]. Outra opção de diagnóstico é uma infusão de catecolaminas. Contudo, em comparação com o teste de stress, existe uma sensibilidade inferior de cerca de 75%, pelo que só deve ser utilizado para fins de diagnóstico em casos excepcionais [10]. O exame electrofisiológico com estimulação programada não tem qualquer valor no diagnóstico do CPVT [2,10]. Como mencionado acima, o ECG de repouso e as técnicas de imagiologia não fornecem valor de diagnóstico. No entanto, os resultados normais destes exames são obrigatórios para se poder fazer o diagnóstico [6].
O diagnóstico diferencial deve incluir outras doenças dos canais iónicos, como a síndrome do QT longo, especialmente o SCD durante a actividade física, como a natação [11]. Um ECG de exercício pode desmascarar o prolongamento do QTc na fase de recuperação, que não é detectável no ECG de repouso, e assim contribuir para a diferenciação do CPVT [12]. Outro diagnóstico diferencial é a síndrome de Andersen-Tawil, que, tal como um subtipo de CPVT, está associada a uma mutação no KCNJ2 e também pode apresentar taquicardia ventricular bidireccional [13]. Para além dos sinais fenotípicos de paralisia periódica e dismorfia das extremidades, que nem sempre estão presentes, o ECG de stress pode também ajudar a diferenciar isto do CPVT. A diferenciação é importante porque os doentes com síndrome de Andersen-Tawil têm um prognóstico mais benigno [13]. Para além das doenças dos canais iónicos, deve-se sempre pensar em doenças estruturais ainda não diagnosticadas, tais como as cardiomiopatias arritmogénicas, hipertróficas, isquémicas ou valvulares, que são muito mais comuns em geral. Outras causas de TV bidireccional são a intoxicação por digoxina ou a miocardite [14,15].
Terapia
O tratamento do CPVT consiste em mudanças de estilo de vida, terapia medicamentosa, estratificação de risco para um CDI até à denervação simpática do coração esquerdo (LKSD). Uma vez que a mortalidade chega a 50% nos doentes gravemente afectados, a terapia adequada é de grande importância [2,16] (Tab. 2) . Além disso, o rastreio familiar deve ser sempre realizado devido à herança autossómica dominante do defeito RYR2.
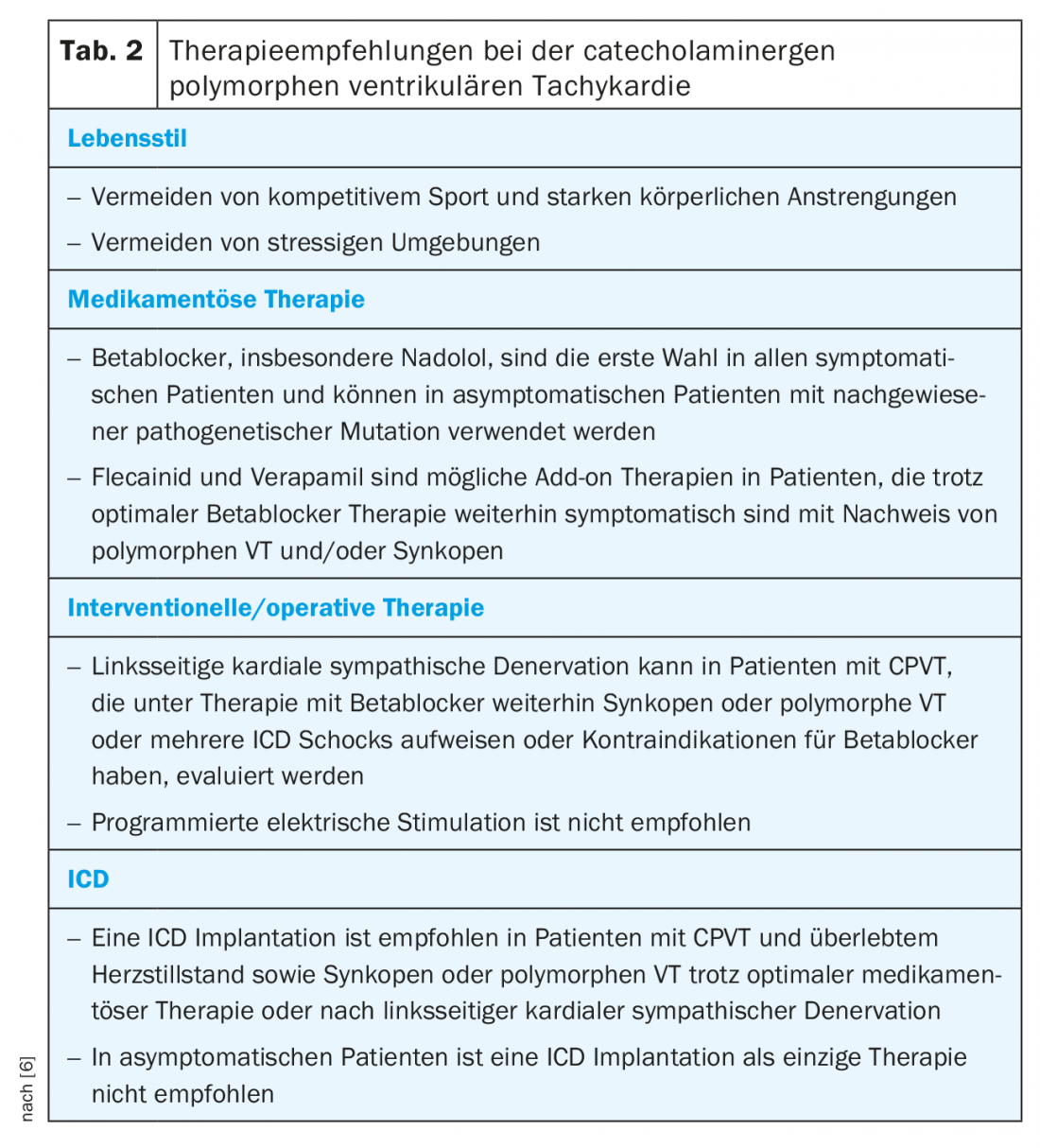
Uma vez que o esforço físico pode desencadear arritmias ventriculares, os pacientes devem evitar o exercício competitivo. Um pequeno estudo foi capaz de mostrar que em pacientes com CPVT adequadamente tratados, o exercício competitivo apenas resultou em mais arritmias sem um aumento da mortalidade [17], mas a transferibilidade para todos os pacientes não é possível. Especialmente ao nadar, deve estar presente um supervisor.
Os beta-bloqueadores são a base da terapia medicamentosa porque, entre outras coisas, impedem que o limite do ritmo cardíaco arritmogénico seja atingido. A maioria dos dados está disponível para o nadolol não-selectivo de longa duração. No entanto, esta preparação já não está disponível em muitos países, tais como a Suíça [16,18,19]. Uma alternativa é o carvedilol, que tem um efeito comprovado no RYR2 [20]. No entanto, faltam dados clínicos em doentes com CPVT. Em princípio, deve ser sempre administrada uma dose completa até à dose máxima tolerável. É importante notar que nas crianças a dose deve ser ajustada ao peso corporal. Uma terapia beta-bloqueadora adequada pode reduzir a taxa de SCD fatal para 6,4% ao longo de 8 anos [19]. Do mesmo modo, os pacientes genótipo-positivos que são detectados por rastreio familiar mas não mostram arritmias no ECG de exercício devem receber terapia com beta-bloqueador [6]. Se as arritmias ou parênteses e as extra-sístoles ventriculares agrupadas continuarem a ser observadas na ergometria do exercício com terapia beta-bloqueadora adequada, deve ser avaliada a terapia adicional com flecainida. Flecainide também mostra um efeito inibitório directo no RYR2 e também foi demonstrado em ensaios clínicos para suprimir as arritmias ventriculares em 76% dos pacientes já tratados com beta-bloqueadores [21,22]. Os bloqueadores dos canais de cálcio parecem ter um benefício adicional como uma terapia alternativa complementar. Pequenos estudos não randomizados mostraram que o verapamil, além dos beta-bloqueadores, reduz ainda mais a ocorrência de arritmias ventriculares [23,24]. Ainda não estão disponíveis ensaios aleatórios maiores de verapamil. As opções terapêuticas ainda não utilizadas clinicamente mas potencialmente promissoras incluem a propafenona, que se revelou promissora em número limitado de doentes [25] e o dantroleno, que demonstrou eficácia num modelo celular [26].
Se as arritmias não forem adequadamente controladas com terapia médica, um LSD pode ser avaliado através da remoção toracoscópica da metade inferior do gânglio estrelado esquerdo e dos gânglios torácicos T2-4. Isto resulta numa diminuição acentuada da secreção de noradrenalina no coração com resultados muito bons em pequenas séries de pacientes com CPVT grave. A complicação mais frequente do LKSD é a síndrome de Horner, na sua maioria transitória, que, no entanto, também pode persistir em 2-3%. Complicações menos comuns são lesões nas estruturas circundantes como a pleura, o nervo frênico, o plexo braquial e os nervos somáticos, resultando em dor de tiro no ombro esquerdo. Como efeito secundário, pode haver falta de suor da mão esquerda e testa esquerda com pele mais quente e seca em comparação com a lateral [27,28].
O implante de CDI só deve ser realizado em pacientes seleccionados que tenham sobrevivido a um CDI ou que continuem a experimentar síncope ou VT polimórfico ou bidireccional, apesar da terapia óptima. Se um LKSD estiver disponível, isto também deve ser realizado antes da implantação [6]. Esta recomendação restritiva existe principalmente porque os choques de CDI per se conduzem à libertação adicional de catecolaminas. Estes podem então provocar mais arritmias e levar a um círculo vicioso, pelo que o CDI deve ser programado com elevados cortes e grandes atrasos antes da entrega do choque [6,29].
Resumo
A CPVT é uma doença extremamente rara que ocorre quase exclusivamente em crianças e adolescentes e tem uma elevada taxa de mortalidade se não for tratada. A terapia com beta-bloqueadores é a intervenção mais importante em termos de prognóstico, juntamente com o ajustamento do estilo de vida. A implantação do CDI está associada a efeitos especiais, potencialmente proarrítmicos, com uma possível descarga de choque, pelo que a indicação na profilaxia primária deve ser feita com cautela. São necessários mais estudos para melhor compreender o quadro clínico e para encontrar novas abordagens terapêuticas.
Mensagens Take-Home
- O CPVT ocorre quase exclusivamente na infância e adolescência em corações estruturalmente normais e tem uma elevada taxa de mortalidade se não for tratado.
- As principais terapias são o ajustamento do estilo de vida e os beta-bloqueadores.
- O implante de CDI só deve ser realizado em pacientes com morte cardíaca súbita, síncope ou taquicardia ventricular polimórfica persistente, apesar da terapia medicamentosa máxima ou após avaliação da denervação simpática do lado esquerdo do coração.
- A entrega de choques por um CDI tem um risco elevado de proarritmia, pelo que tempos de detecção longos e frequências de intervenção elevadas devem ser programados para o CDI.
Literatura:
- Reid DS, et al: Taquicardia bidireccional numa criança. Um estudo utilizando a sua electrografia de feixe. Br Coração J. 1975; 37:339-344.
- Leenhardt A, et al: Taquicardia ventricular polimórfica catecolaminérgica em crianças. Um acompanhamento de 7 anos de 21 pacientes. Circulação. 1995; 91: 1512-1519.
- Priori SG, et al: Caracterização clínica e molecular de pacientes com taquicardia ventricular polimórfica catecolaminérgica. Circulação. 2002; 106: 69-74.
- Leenhardt Antoine, Denjoy Isabelle, Guicheney Pascale. Taquicardia Ventricular Polimórfica Catecolaminérgica. Electrofisiol de Arritmia Circ. 2012; 5: 1044-1052.
- Pérez-Riera AR, et al: Catecolaminérgico polimórfico de taquicardia ventricular polimórfica, uma actualização. Ann Noninvasive Electrocardiol Off J Int Soc Holter Noninvasive Electrocardiol Inc. 2018;23: e12512.
- Declaração de Consenso de Peritos HRS/EHRA/APHRS sobre o Diagnóstico e Gestão de Pacientes com Síndromes Hereditárias de Arritmia Primária. 2013; 69.
- van der Werf C, Wilde AAM: Taquicardia ventricular polimórfica catecolaminérgica: do banco para a cabeceira do leito. Coração. 2013; 99: 497-504.
- Svendsen JH, Geelen P, EHRA Comité de Iniciativa Científica: Rastreio e gestão de possíveis síndromes arritmogénicas (doenças do canal de canalização/ião). Eur Eur Arritm Arritm Card Electrophysiol J Grupos de trabalho Cartão de Arritm Arritm Card Electrophysiol Eur Soc Cardiol. 2010; 12: 741-742.
- Sy RW, et al: Caracterização da arritmia e resultados a longo prazo em taquicardia ventricular polimórfica catecolaminérgica. Ritmo cardíaco. 2011; 8:864-871.
- Sumitomo N, et al.: Taquicardia ventricular polimórfica catecolaminérgica: características electrocardiográficas e estratégias terapêuticas óptimas para prevenir a morte súbita. Coração. 2003;89: 66-70.
- Tester DJ, et al: Afogamentos inexplicáveis e as canalizações cardíacas: uma série de autópsias moleculares. Mayo Clin Proc. 2011; 86: 941-947.
- Sy RW, et al: Derivação e validação de um algoritmo simples baseado em exercício para a previsão de testes genéticos em familiares de sondas LQTS. Circulação. 2011; 124: 2187-2194.
- Inoue YY, et al.: Diferentes respostas ao exercício entre a síndrome de Andersen-Tawil e a taquicardia ventricular polimórfica catecolaminérgica. Eur Eur Pacing Arrhythm Card Electrophysiol J Grupos de trabalho Card Pacing Arrhythm Card Cell Electrophysiol Eur Soc Cardiol. 2018; 20: 1675-1682.
- Berte B, et al: Taquicardia ventricular bidireccional em miocardite fulminante. Eur Eur Pacing Arrhythm Card Electrophysiol J Grupos de trabalho Card Pacing Arrhythm Card Cell Electrophysiol Eur Soc Cardiol. 2008; 10: 767-768.
- Valent S, Kelly P: Imagens em medicina clínica. Taquicardia ventricular bidireccional induzida por digoxina. N Engl J Med. 1997; 336: 550.
- Hayashi M, et al.: Incidência e factores de risco de eventos arrítmicos em taquicardia ventricular polimórfica catecolaminérgica. Circulação. 2009; 119: 2426-2434.
- Ostby SA, et al: Competitive Sports Participation in Patients With Catecholaminergic Polymorphic Ventricular Tachycardia: A Single Center’s Early Experience. JACC Clin Electrophysiol. 2016; 2: 253-262.
- Leren IS, et al: Nadolol diminui a incidência e a gravidade das arritmias ventriculares durante o teste de esforço de exercício em comparação com β1-selectivo β-bloqueadores em doentes com taquicardia ventricular polimórfica catecolaminérgica. Ritmo cardíaco. 2016; 13: 433-440.
- van der Werf C, Zwinderman AH, Wilde AAM: Abordagem terapêutica para doentes com taquicardia ventricular polimórfica catecolaminérgica: estado da arte e desenvolvimentos futuros. Eur Eur Arritm Arritm Card Electrophysiol J Grupos de trabalho Cartão de Arritm Arritm Card Electrophysiol Eur Soc Cardiol. 2012; 14:175-183.
- Zhou Q, et al: O Carvedilol e os seus novos análogos suprimem a libertação de Ca2+ induzida pela sobrecarga arritmogénica do armazém. Nat Med. 2011; 17: 1003-1009.
- van der Werf C, et al: A terapia Flecainide reduz arritmias ventriculares induzidas pelo exercício em doentes com taquicardia ventricular polimórfica catecolaminérgica. J Am Coll Cardiol. 2011; 57: 2244-2254.
- Watanabe H, et al: Flecainide previne taquicardia ventricular polimórfica catecolaminérgica em ratos e humanos. Nat Med. 2009; 15: 380-383.
- Rosso R, et al.: Bloqueadores dos canais de cálcio e beta-bloqueadores versus beta-bloqueadores apenas para prevenir arritmias induzidas pelo exercício em taquicardia ventricular polimórfica catecolaminérgica. Ritmo cardíaco. 2007; 4: 1149-1154.
- Swan H, et al: O antagonismo do canal de cálcio reduz as arritmias ventriculares induzidas pelo exercício em doentes com taquicardia ventricular polimórfica catecolaminérgica com mutações de RyR2. J Cardiovasc Electrophysiol. 2005; 16: 162-166.
- Hwang HS, et al: A inibição dos canais de libertação de Ca2+ cardíaca (RyR2) determina a eficácia dos medicamentos antiarrítmicos classe I na taquicardia ventricular polimórfica catecolaminérgica. Circ Arrhythm Electrophysiol. 2011; 4: 128-135.
- Jung CB, et al: Dantrolene salva defeito RYR2 arritmogénico num modelo de células estaminais de taquicardia ventricular polimórfica catecolaminérgica específica de um paciente. EMBO Mol Med. 2012; 4: 180-191.
- Odero A, et al: Desnervação simpática cardíaca esquerda para a prevenção de arritmias que ameaçam a vida: a abordagem cirúrgica supraclavicular à simpatectomia cervicotorácica. Ritmo cardíaco. 2010; 7: 1161-1165.
- Wilde AAM, et al: Desnervação simpática do coração esquerdo para taquicardia ventricular polimórfica catecolaminérgica. N Engl J Med. 2008; 358: 2024-2029.
- Mohamed U, et al: morte cardíaca súbita apesar de um cardioversor-desfibrilador implantável numa jovem fêmea com taquicardia ventricular catecolaminérgica. Ritmo cardíaco. 2006; 3: 1486-1489.
CARDIOVASC 2019; 18(2): 12-15