As vasculites sistémicas são síndromes inflamatórias dos vasos sanguíneos, que podem causar um espectro muito amplo de sintomas dependendo do calibre do vaso afectado e da localização. Os vasculídios primários mais comuns em adultos são descritos com as opções actuais de terapia medicamentosa.
As vasculites sistémicas são síndromes inflamatórias dos vasos sanguíneos, que podem causar um espectro muito amplo de sintomas dependendo do calibre do vaso afectado e da localização. Dependendo da localização, um ataque vasculítico de pequenos vasos, ou seja de capilares, arteríolas e vênulas, pode manifestar-se, por exemplo, como púrpura palpável da pele, glomerulonefrite necrotizante rápida com insuficiência renal, hemorragias pulmonares, hemorragias nasais, esclerites ou como opacidade cerebral. Se as artérias médias e grandes forem afectadas, então existe o risco de enfartes de tecidos, aneurismas, hemorragias e tromboses. Embora tenham sido feitos progressos significativos no tratamento da vasculite sistémica ao longo dos últimos 40 anos, a mortalidade continua a aumentar significativamente em comparação com a população em geral [1]. Os vasculídios primários mais comuns em adultos são descritos abaixo, juntamente com as opções actuais de terapia medicamentosa.
Classificação dos vasculídios sistémicos
O grupo de vascultídeos “primários” sistémicos compreende síndromes independentes de doenças “idiopáticas”, enquanto que os vascultídeos “secundários” ocorrem em ligação com doenças pré-existentes. Exemplos de vasculites secundárias são a vasculite crioglobulinémica associada à hepatite C e a vasculite na artrite reumatóide seropositiva de longa data.
A classificação dos vascultídeos primários sistémicos de acordo com a classificação Chapel-Hill é baseada no calibre do recipiente afectado [2]. (Resumo1). As vasculites de grandes vasos que afectam a aorta e/ou os seus ramos incluem a arterite Takayasu, que ocorre principalmente em mulheres jovens asiáticas, e a arterite de células gigantes (RZA; sinónimo: arterite temporal), que normalmente não se manifesta antes dos 50 anos de idade. A síndrome de Kawasaki, que afecta as crianças, e a muito rara panarterite nodosa afectam particularmente as artérias de média dimensão. Os vascultídeos primários dos pequenos vasos estão divididos em dois grupos principais: Os ANCA (anticorpos anti-neutrofílicos citoplasmáticos)-associados aos vascultídeos (AAV) e os ANCA-negativos vascultídeos. AAVs incluem granulomatose com poliangite (GPA; antiga doença de Wegener); poliangite microscópica (MPA); granulomatose eosinofílica com poliangite (EGPA; antiga síndrome de Churg-Strauss); e glomerulonefrite necrotizante focal (FNGN). Uma série de vasculites mediadas por complexos imunológicos são atribuídas aos vasculites primários primários de pequenos vasos negativos ANCA, por exemplo a vasculite IgA (Henoch-Schönlein), que ocorre principalmente em crianças, e a vasculite crioglobulina essencial.

Os vascultídeos secundários afectam geralmente pequenos vasos (capilares, arteríolas, vênulas) e, tal como os vascultídeos ANCA-negativos de pequenos vasos, são geralmente causados por complexos imunitários com activação do sistema complemento. O tratamento da vasculite secundária está sempre relacionado com a doença subjacente e envolve muito frequentemente a utilização a curto prazo de doses mais elevadas de glicocorticóides e, em alguns casos (por exemplo, a vasculite crioglobulina induzida por vírus), o esgotamento terapêutico das células CD20+ B [3]. A doença de Behçet ocupa uma certa posição especial entre as síndromes de vasculite, na qual todos os calibres de vasos podem ser afectados.
Em todos os vasculípedos, o primeiro objectivo do tratamento é conseguir uma remissão clínica tão rápida quanto possível, o que se chama indução de remissão. Segue-se a fase de manutenção da remissão, que pode durar vários anos, dependendo do tipo de vasculite.
Arterite de células gigantes (RZA)
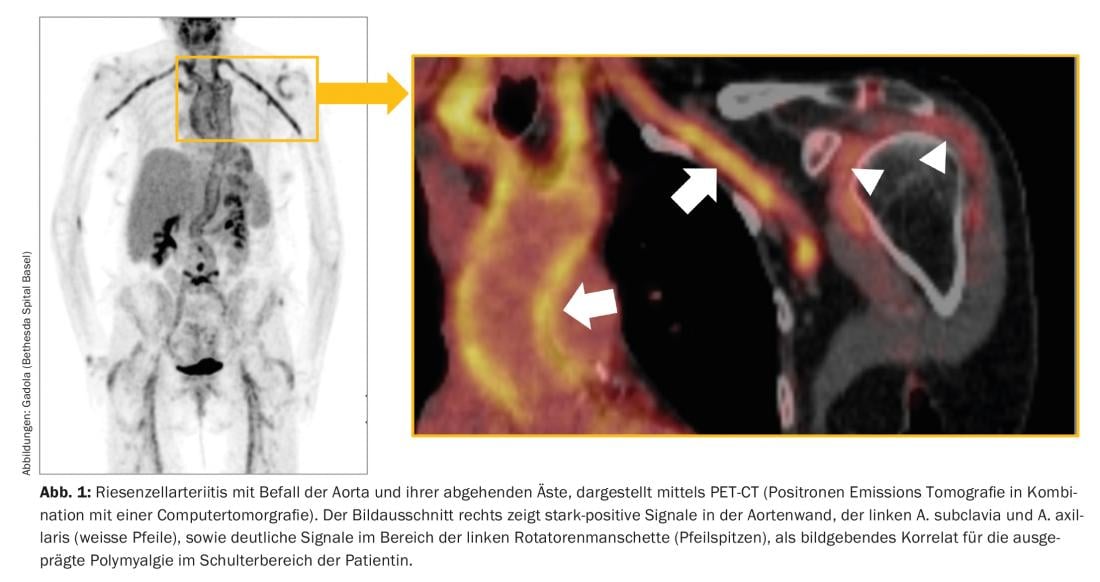
O RZA, também conhecido como arterite temporalis, é a vasculite mais comum nas nossas latitudes e ocorre frequentemente após os 60 anos de idade, e na grande maioria dos casos após os 50 anos de idade. Afecta a aorta e as grandes e médias artérias que dela se ramificam (Fig. 1). Os sintomas típicos são dores de cabeça novas, frequentemente “nevrálgicas”, síndrome polimialégica, sintomatologia B com queda de desempenho, exaustão e suores nocturnos, sintomas de claudicação das extremidades, língua e/ou músculos mastigatórios, bem como distúrbios visuais. O RZA é temido principalmente por causa do perigo de cegueira aguda, que é geralmente irreversível. Em termos laboratoriais, uma reacção de fase aguda pronunciada é típica – mas não obrigatória – com reacção de sedimentação acentuadamente acelerada (BSR) e elevação da proteína C-reactiva (CRP).
Os glicocorticóides são os medicamentos de primeira linha para o RZA e normalmente resultam numa melhoria dramática dos sintomas subjectivos em 24 horas. Se houver suspeita de envolvimento ocular, por exemplo, no caso de perturbações transitórias da acuidade visual, e cegueira potencial iminente, doses elevadas, por exemplo 1 g de metilprednisolona em três dias consecutivos, são administradas por via intravenosa. Nos outros casos, uma dose diária inicial de 40-60 mg é suficiente. A dose é ajustada de acordo com os valores da clínica e do laboratório. Enquanto a dose de prednisona não puder ser reduzida abaixo de 20 mg/dia, a profilaxia antibiótica com sulfometoxazol-trimetoprim (por exemplo Cotrim-CT 800/160 mg; Bactrim forte®) 3×1/semana deve ser administrada para prevenir infecções oportunistas, especialmente as causadas por Pneumocystis jirovecii.
No passado, foram utilizadas doses de prednisona de 1 mg/kg de peso corporal durante um ano inteiro no RZA, como noutras vasculites, com ocorrência frequente de efeitos secundários graves. No entanto, com a ajuda de medicamentos básicos anti-reumáticos com esteróides (medicamentos anti-reumáticos modificadores de doenças, DMARD), a dose de prednisona pode ser reduzida para abaixo da chamada dose limite de esteróides de 7,5 mg de prednisona muito mais cedo. O metotrexato (MTX), em particular, provou ser um bom DMARD para o RZA. O início da acção é atrasado com o MTX, como com todos os DMARD, e ocorre aproximadamente 4-6 semanas após atingir a dose efectiva.
O metotrexato deve ser sempre administrado parenteralmente, isto é, subcutaneamente (s.c.) uma vez por semana, para o tratamento de vasculites. Se bem tolerada, a dose pode ser aumentada para um máximo de 0,3 mg/kgKG por semana. O BSR e o CRP também servem como valiosos biomarcadores de progressão da actividade inflamatória em tratamento com prednisona e DMARD. Para reduzir os efeitos secundários tóxicos do MTX, o ácido fólico, por exemplo 10 mg/semana, deve ser sempre tomado concomitantemente no dia seguinte à injecção do metotrexato. O MTX nunca deve ser combinado com sulfometoxazol-trimetoprim, caso contrário pode ocorrer uma severa mielossupressão. Por conseguinte, só utilizamos MTX após a dose de prednisona ter sido reduzida para menos de 20 mg/dia.
Antes de iniciar a terapia básica com MTX ou outros DMARD, deve ser realizada uma radiografia de tórax para excluir infecção crónica ou fibrose pulmonar, um hemograma diferencial, valores hepáticos e renais, e testes serológicos para a hepatite B, hepatite C e VIH. Durante os primeiros 3 meses sob metotrexato, são recomendados controlos mensais dos valores hepáticos e renais e do hemograma; depois disso, estes controlos podem ser efectuados a intervalos mais longos de 8 a 12 semanas, se necessário. Recomendações detalhadas para a utilização de DMARD em doenças reumatológicas podem ser encontradas nos portais de Internet das sociedades reumatológicas [p. ex. 4].
Desde 2017 (zona UE) resp. 2018 (Suíça), o receptor anti-interleucina-6 (anti-IL6R) de anticorpos tocilizumab (Actemra®) é aprovado para o tratamento do RZA. Com tocilizumab, a dose de glicocorticóides pode ser reduzida muito mais rapidamente, mesmo sem metotrexato, com bom sucesso clínico [5,6]. A terapia com tocilizumab deve ser realizada durante pelo menos 1 ano, pois de outra forma as recidivas são frequentes [7]. O BSR e o CRP são suprimidos sob tocilizumab e, portanto, não são úteis como parâmetros de progressão da actividade da doença. Dados recentes sugerem que a dose de glicocorticóide sob tocilizumabe pode ser interrompida muito rapidamente, ou seja, dentro de algumas semanas [8]; no entanto, são necessários mais estudos antes de se poderem fazer recomendações claras. Como os glicocorticóides no RZA não podem actualmente ser reduzidos abaixo da dose limite durante um período de tempo mais longo (>3 meses), recomenda-se o rápido início da terapia anti-reabsortiva, por exemplo com alendronato, para prevenir a reabsorção óssea adquirida com esteróides ou a perda óssea. de osteoporose.
Arterite Takayasu (TA)
Semelhante ao RZA, TA afecta a aorta e as grandes artérias que dela derivam. Ao contrário do RZA, esta vasculite ocorre em 80-90% dos casos em mulheres, com início entre os 10 e 40 anos de idade . A AT progride em episódios, e tipicamente manifesta-se com sintomas constitucionais, artralgias, e – caracteristicamente – dores de pressão carotídea marcadas (em 10-30%). No decurso da doença, pode ocorrer oclusão vascular, hipertensão renovascular grave, (Takayasu) retinopatia, e aneurismas da aorta com ou sem insuficiência da válvula aórtica.
Quando a TA é diagnosticada, os glicocorticóides são utilizados em primeira instância. Os DMARD modificadores de esteróides mais frequentemente utilizados em TA são o metotrexato s.c. (como no RZA) ou azatioprina p.o. (até 2 mg/kgKG). As alternativas são o micofenolato (1,5 g-3 g/dia p.o.) e a leflunomida (20 mg/dia p.o.). Em casos de intolerância ao metotrexato ou DMARD oral, são utilizados casos resistentes e graves, bloqueadores TNFalpha- (por exemplo, etanercept ou infliximab) ou outros biológicos (por exemplo, tocilizumab, abatacept, ustekinu-mab), mas estes ainda não estão aprovados para esta indicação [9].
Vasculídios associados à ANCA (AAV)
GPA (antiga doença de Wegener)
GPA é a vasculite primária mais comum nas nossas latitudes, com uma distribuição de género quase equilibrada e uma idade típica de manifestação entre 40-60 anos. Ano de vida. É feita uma distinção entre uma “fase inicial” não vasculítica e granulomatosa (localizada) e uma “fase de generalização” sistémica e vasculítica, que pode ocorrer sequencialmente (fase inicial ‘ fase de generalização) ou simultaneamente. Embora exista hoje uma vasta gama de terapias eficazes para o tratamento da vasculite em pequenos vasos, o tratamento das manifestações inflamatórias granulomatosas agressivas é frequentemente um grande desafio [10,11].
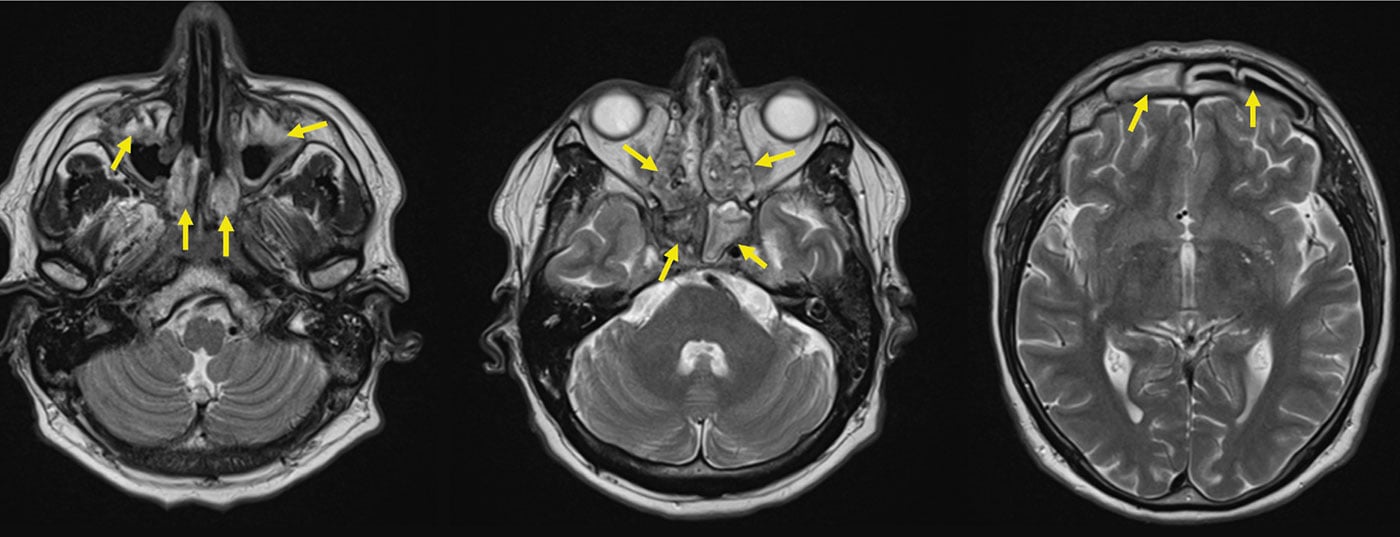
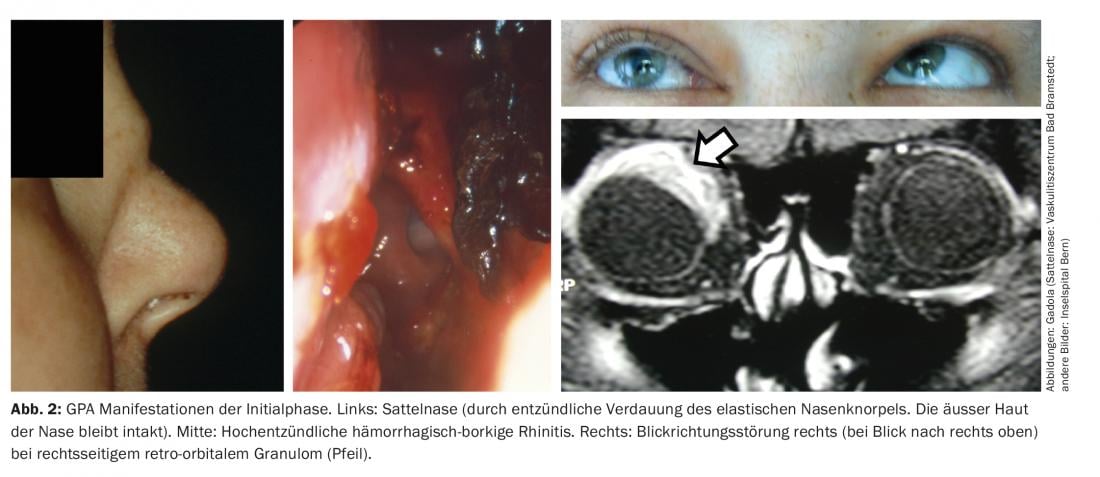
Fase inicial do AGP (AGP “localizado”). A fase inicial da GPA manifesta-se tipicamente no ouvido, nariz e garganta e nas vias respiratórias, por exemplo sob a forma de rinite borky hemorrágica crónica, sinusite ou mastoidite resistente à terapia. Num percurso agressivo, pode ocorrer um nariz de sela, devido à destruição da cartilagem nasal elástica, formação de fístulas na órbita ou em direcção à face, granulomas retro-orbitais com direcção da visão prejudicada (diplopia), e paquimeningite granulomatosa (Fig. 2). Estes dois últimos devem-se provavelmente a uma inflamação granulomatosa originada pelos seios nasais por continuitatem [11]. ANCA com especificidade para proteinase-3 (PR3-ANCA) ou, mais raramente, mieloperoxidase (MPO-ANCA) são detectáveis em apenas cerca de 50% dos pacientes com GPA durante a fase inicial.
Para o tratamento da fase inicial “pura” sem vasculite sistémica concomitante, são utilizadas diferentes combinações de medicamentos, dependendo da gravidade dos sintomas. Em casos mais leves, o sulfomethoxazol-trimetoprim (T/S) é utilizado com ou sem prednisona de baixa dose (10 mg/dia). A utilização do T/S remonta a uma observação empírica nos anos 70 por Richard Deremee na Mayo Clinic [12]. Nos anos 90, foi demonstrada uma associação entre a colonização nasal crónica por Staphylococcus aureus e a actividade da doença na GPA [13]. Desde então, o tratamento antibiótico intra-nasal tópico com mupirocina também tem sido utilizado, mas sem sucesso retumbante. Uma análise recente do microbioma endonasal na GPA mostrou uma associação interessante da actividade da doença na GPA com Corynebacterium tuberculostearicum [14]. Este agente patogénico é um agente patogénico importante em outras doenças granulomatosas [15]. Corynebacterium tuberculostearicum é resistente à maioria dos antibióticos [16], o que pode explicar o sucesso moderado da T/S e da mupirocina.
No curso mais agressivo da fase inicial, o metotrexato (sempre sem T/S, como de resto a toxicidade combinada da medula óssea) é utilizado principalmente em combinação com a prednisona. Em casos refractários, grandes granulomas pulmonares e também granulomas retro-orbitais, a terapia com anticorpos anti-CD20 (Rituximab) pode ser eficaz [17]. Os granulomas em GPA têm uma alta densidade de células CD20+ B e são vistos como um possível site de origem da produção de ANCA [18,19]. Uma manifestação particular da GPA durante a fase inicial é a estenose traqueal inflamatória fibrosante com dispneia e estridor inspiratório, que pode requerer tratamento com infiltração local de glicocorticóides e dilatação por balão.
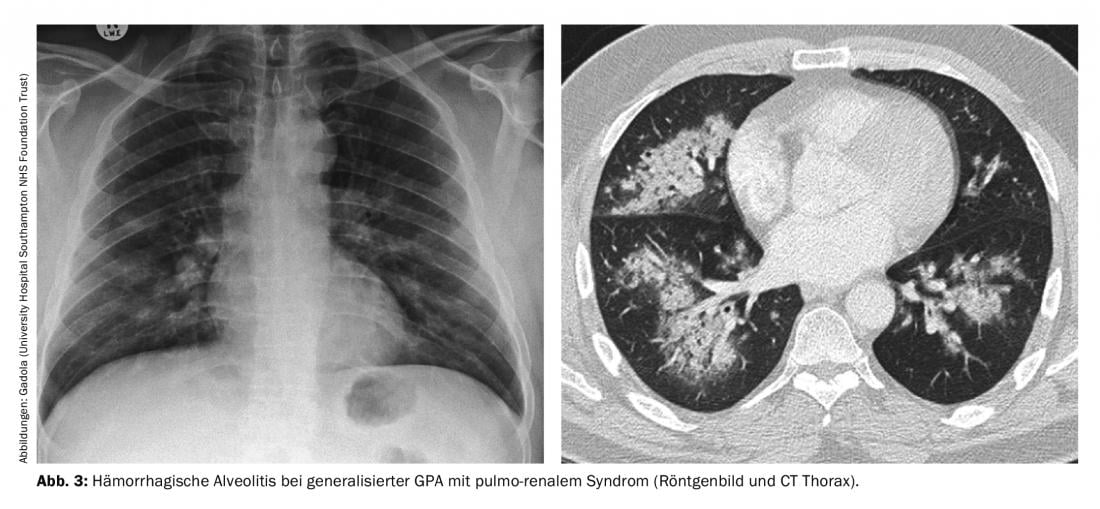
Fase de Generalisaton do GPA (ANCA em >98%). A vasculite sistémica de pequenos vasos em GPA, na qual PR3-ANCA ou MPO-ANCA pode praticamente sempre ser detectada no soro, pode afectar todos os órgãos. A infecção dos rins conduz tipicamente a uma rápida glomerulonefrite “pauci-imune” (RPGN) progressiva e necrosante, que, se não for tratada, pode conduzir rapidamente a uma insuficiência renal grave que requer diálise. A síndrome pulmonar, ou hipertensão pulmonar, é particularmente temida. a ocorrência combinada de RPGN com alveolite hemorrágica (Fig. 3), que tem uma mortalidade elevada. Outras manifestações típicas da fase de generalização são uma clara sintomatologia B, esclerose – típica é a dolorosa escleromalácia nodular – que pode levar à escleromalácia, a mononeurite múltipla frequentemente muito dolorosa, encefalite com opacificação, púrpura palpável, e poliartrite nãoerosiva. Os seguintes princípios de tratamento para GPA generalizada podem ser aplicados à MPA e à FNGN, e parcialmente à EGPA.
Indução de remissão em GPA generalizada. Dependendo da gravidade da manifestação, estão disponíveis diferentes modos de acção para induzir a remissão. A regra principal na GPA generalizada é que a actividade inflamatória deve ser sempre controlada tão rápida e minuciosamente quanto possível.
No caso de GPA sistémica não-renal e de outro modo não ameaçadora para os órgãos, a terapia de indução de remissão pode ser iniciada com metotrexato mais prednisona ou com pulsos intravenosos (i.v.) de ciclofosfamida (por exemplo, 4 pulsos de 10 mg/kgKG cada um em intervalos de 3 semanas) mais prednisona, desde que seja possível monitorizar o progresso clínico de perto pelo menos uma vez por semana. No caso de manifestações renais e outras manifestações ameaçadoras para os órgãos, além da prednisona, a ciclofosfamida é utilizada principalmente por via oral (começando com 2 mg/kgKG/dia) ou como pulso i.v., ou rituximab (2 infusões i.v. com 1g de rituximab cada uma em intervalos de 14 dias) em combinação com a prednisona. A escolha entre o tratamento oral mais intensivo com ciclofosfamida e a terapia de pulso requer sempre uma cuidadosa consideração da relação risco-benefício por parte do especialista experiente com a mesma. Sob ciclofosfamida, os leucócitos em particular, e especialmente os granulócitos neutrófilos, que são tipicamente aumentados significativamente na vasculite GPA activa, devem ser determinados regularmente. Os leucócitos devem atingir nadir dentro de 8-10 dias sob ciclofosfamida, o que deve ser sempre determinado. Se os leucócitos não caírem, então isto indica actividade persistente da doença, o que pode exigir o aumento da dose “adaptada aos leucócitos” para 2,5-3 mg/kgKG. Sob ciclofosfamida, podem ocorrer infecções oportunistas graves logo após o início da terapia, quando ocorre leucopenia, especialmente a reactivação do citomegalovírus (CMV). Os efeitos secundários tóxicos típicos da bexiga relacionados com a dose são cistite hemorrágica e carcinomas da bexiga, que ocorrem após uma dose cumulativa de pelo menos 25 g (e uma média de 100 g) de ciclofosfamida [20], bem como síndrome mielodisplásica (MDS). A probabilidade de complicações vesicais pode ser reduzida por uma boa hidratação durante todo o período de tratamento e pelo uso adicional de Mesna (Uromitexan), que liga e neutraliza a acroleína do metabolito da ciclofosfamida.
O esgotamento duradouro das células B com imunodeficiência humoral secundária e infecções respiratórias graves frequentes podem ocorrer com rituximab, especialmente em doentes que tenham recebido previamente ciclofosfamida [21].
Em cursos particularmente severos e/ou refractários de GPA sistémica, são por vezes utilizadas medidas e fármacos adicionais, incluindo plasmaferese, administração de imunoglobulina intravenosa, micofenolato mofetil (MMF), o anticorpo anti-CD52 alemtuzumab, a globulina anti-timócitos, a 15-deoxi-spergualina – um inibidor da diferenciação celular – e o transplante de células estaminais hematopoiéticas. O sucesso destes tratamentos intensificados não está bem estabelecido e a sua utilização deve ser restrita a centros experientes.
Um novo mecanismo de acção muito promissor para o tratamento de GPA generalizada, bem como de outros AAV, é a inibição do sistema complemento. Avacopan, um antagonista oral do receptor C5a, mostrou resultados muito promissores nos ensaios da fase 3, pelo que a probabilidade de aprovação num futuro próximo é elevada. Nos doentes com GPA generalizada que receberam Avacopan para além da terapia padrão (com ciclofosfamida ou rituximab), os glicocorticóides poderiam ser descontinuados muito rapidamente. Curiosamente, a função renal em doentes tratados com Avacopan mostrou uma melhoria contínua durante o período de tratamento de 52 semanas [22].
Um medicamento existente para inibir o sistema complemento é o anti-C5 anticorpo eculizumab (Soliris®), que tem sido utilizado esporadicamente para a progressão agressiva do AAV [por exemplo 24]. Um ensaio de fase 2 de eculizumabe em ANCA vasculite foi infelizmente retirado antes da inscrição dos pacientes (NCT01275287). Outro medicamento, o Iptacopan (LNP023), que inibe a activação eficiente do sistema complemento através do factor B, recebeu recentemente aprovação para o tratamento da glomerulopatia C3 (C3G), e é bem possível que este medicamento também venha a ser utilizado em AAV no futuro.
Manutenção de remissão em AGP generalizada. Após a remissão clínica ter sido alcançada, a terapia de manutenção da remissão deve salvaguardar a actividade inflamatória da AAV mesmo com pequenas doses de prednisona ou mesmo sem glucocorticoides concomitantes. Desde 1995, o Grupo Europeu de Estudos sobre Vasculite (EUVAS) tem conduzido um grande número de estudos de intervenção clínica na AAV. MTX, azatioprina ou rituximab são recomendados para manter a remissão em GPA e outros AAV. O Rituximab é possivelmente a droga mais eficaz dos três, embora a dosagem e intervalo de dose óptimos do rituximab em AAV ainda esteja em discussão. Alguns peritos recomendam um intervalo fixo de 6 meses de 1g de rituximab [24], enquanto um estudo comparativo francês de rituximab contra a azatioprina mostrou bons resultados para um intervalo de 6-12 meses com doses de 500mg [25]. Com rituximab, como com ciclofosfamida, os níveis de PR3 ou MPO-ANCA caem abaixo do limite de detecção na maioria dos casos, correlacionando-se com a remissão clínica da vasculite. Nestes pacientes, na nossa experiência, o intervalo rituximab pode ser ajustado ao ressalto em títulos ANCA, prolongando o intervalo para mais de 12 meses em alguns casos.
Outros AAV (MPA, FNGN, EGPA)
MPA e FNGN
Ao contrário do GPA, os granulomas não ocorrem em MPA e FNGN. Estas AAV são ligeiramente mais frequentemente associadas à MPO-ANCA do que à PR3-ANCA, mas isto não importa para o tratamento. Ambas as AAV manifestam-se tipicamente como “pauci-imune”, glomerulonefrite progressiva rápida (RPGN). O termo “pauci-imune” refere-se à fraca detectabilidade dos depósitos de imunoglobulina no exame imuno-histoquímico das biópsias renais. Outras manifestações típicas da MPA são a mononeurite múltipla dolorosa e a alveolite, que podem levar à fibrose pulmonar com um aumento significativo da mortalidade em cerca de um terço dos doentes [26]. O tratamento de MPA e FNGN é análogo aos princípios de tratamento acima descritos para GPA generalizado.
EGPA (anteriormente: síndrome de Churg-Strauss/vasculite)
A EGPA pode ser descrita como a “contraparte atópica” da GPA. Ocorre em doentes com sintomas atópicos que estão presentes há anos a décadas, especialmente asma e/ou pansinusite crónica (Fig. 4) com pólipos nasais. Durante a fase inicial da EGPA, o granuloma é encontrado no tecido afectado, semelhante à GPA. Em contraste com a GPA, contudo, estes granulomas não são densamente intercalados com granulócitos neutrófilos, mas com granulócitos eosinófilos. No sangue periférico dos doentes com EGPA, é encontrada uma eosinofilia de forma análoga. A EGPA difere clinicamente da GPA em aspectos importantes. A ocorrência de RPGN é com aproximadamente 15-20% mais rara do que com a GPA [27], mas como com a GPA estritamente associada com a presença de ANCA. As manifestações neurológicas, especialmente uma mononeurite multiplex frequentemente muito dolorosa [28], ocorrem em mais de metade dos pacientes. As manifestações mais temidas da EGPA são as manifestações cardíacas, por exemplo, vasculite das artérias coronárias, que ocorrem em cerca de 40% dos doentes e são a causa mais comum de morte na EGPA [29,30].
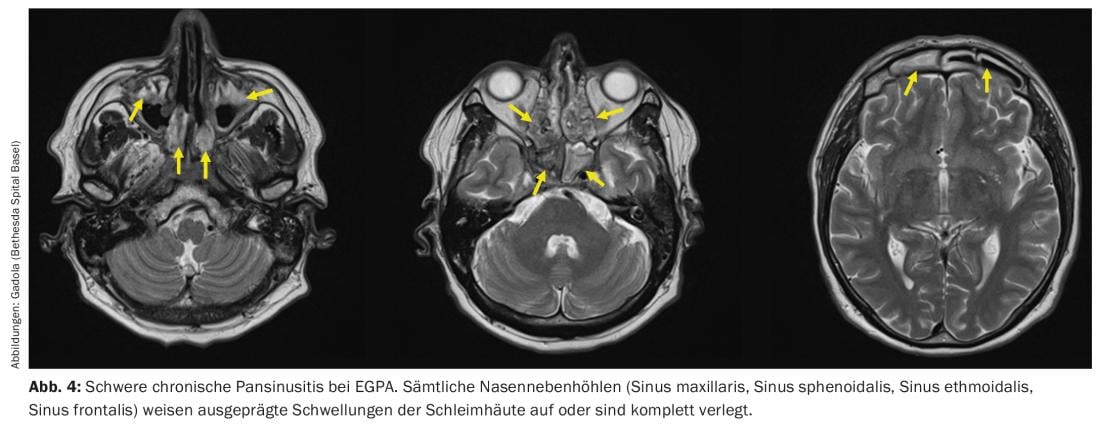
A terapia da EGPA é muito semelhante à da GPA generalizada, e os medicamentos mais comuns na EGPA, como na GPA, são glucocorticóides, MTX, ciclofosfamida e rituximab. As altas doses de glucocorticoides causam muito rapidamente uma queda drástica dos eosinófilos e uma melhoria clínica. No entanto, tal como com a GPA, a terapia com esteróides deve ser reduzida rapidamente e para a dose limite de prednisona no prazo de 3 meses para reduzir o risco de infecções graves das vias aéreas. No caso de EGPA recorrente ou resistente à terapia, estão também disponíveis interferon-alpha (3×/semana a administração diária s.c.) e o anticorpo anti-IL5 mepolizumab (Nucala®, 300 mg s.c. a cada 4 semanas), que também é aprovado para esta indicação. Com uma terapia eficaz, a eosinofilia deve desaparecer e o título de ANCA deve cair.
Vasculite em pequenos vasos não associados àANCA
Vasculite crioglobulinémica essencial
Esta vasculite de pequeno vaso é causada pela activação in situ da cascata complementar nas paredes do vaso após a deposição de crioglobulinas de tipo II (menos comumente de tipo III). Manifesta-se principalmente na pele com vasculite urticaria ou púrpura palpável (Fig. 5) . O envolvimento renal apresenta-se normalmente com complexo imunitário semelhante ao lúpus e glomerulonefrite membranoproliferativa mediada por complementos. Outros sintomas típicos são mialgias e artralgias, bem como a polineuropatia. Durante as recaídas, os complementos C4 (“sempre”) e C3 (“frequentemente”) são reduzidos no soro.
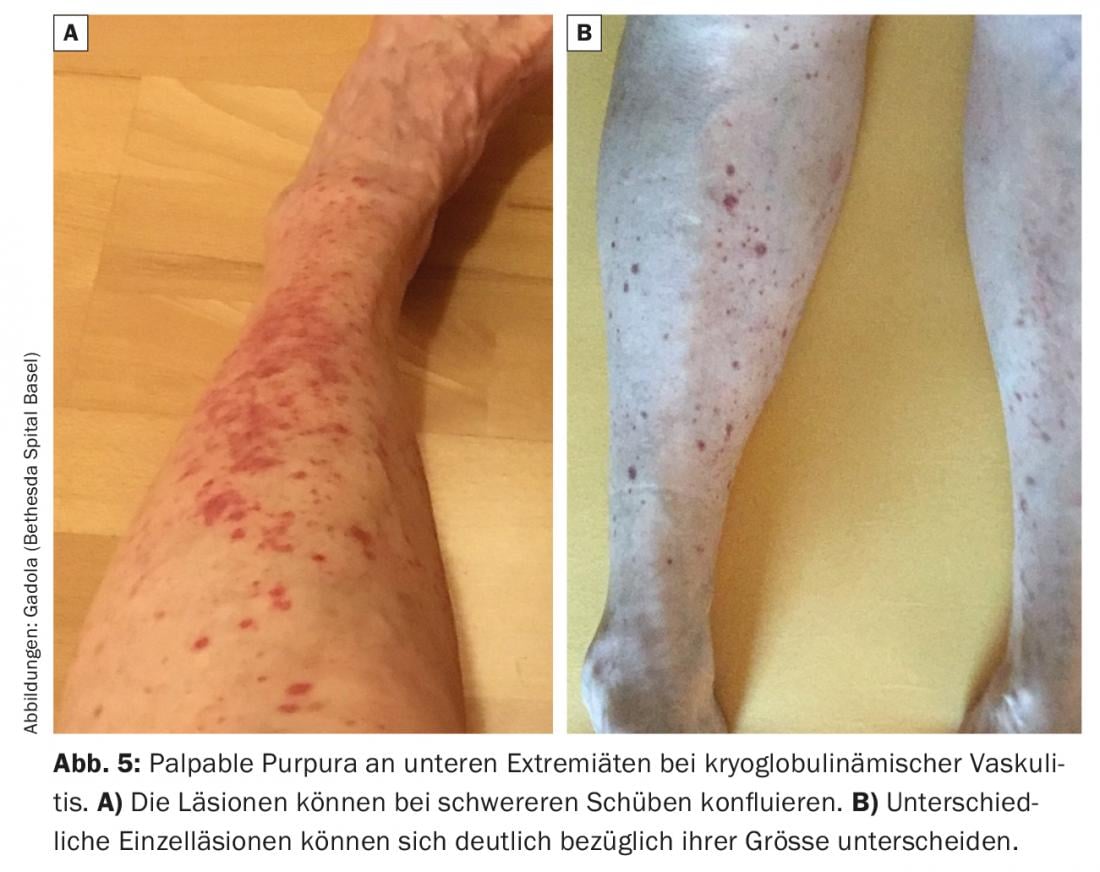
A terapia começa com glicocorticóides e é complementada com DMARD (MTX ou azatioprina), se necessário. Nos casos refractários, o rituximab demonstrou ser muito eficaz e, nos casos agudos graves com grave envolvimento renal, é utilizada a ciclofosfamida [31].
Mais comuns que a vasculite crioglobulinaémica essencial, na qual a etiologia é por definição desconhecida, são a vasculite crioglobulinaémica associada ao HCV (crioglobulinas tipo II ou tipo III no soro) e episódios de vasculite crioglobulinaémica na neoplasia das células B (crioglobulinas tipo I ou tipo III). Aqui, o tratamento da doença subjacente está principalmente em primeiro plano, bem como o tratamento de esgotamento das células B com rituximab em casos graves.
Vasculite leucocitocítica
Normalmente, a vasculite leucocitocítica mediada por complexos imunitários está estritamente confinada aos capilares e vênulas da derme e manifesta-se com púrpura pruriginosa a púrpura dolorosa palpável das extremidades inferiores. Se outros órgãos forem afectados para além da pele, deve sempre procurar-se outra vasculite primária ou secundária de pequenos vasos. Vasculite leucocitocítica confinada à pele. é frequentemente auto-limitada; no entanto, também podem ocorrer cursos severos recorrentes ou crónicos, que são tratados com glucocorticóides e, se necessário, com vários DMARD; em casos particularmente severos, também com ciclofosfamida.
Outras vasculites “idiopáticas” (e portanto “primárias”) de pequenos vasos do adulto são vasculites hipocomplementais hipocomplementais urticárias (HUV), vasculites IgA (Henoch-Schönlein) e vasculites anti-GBM (GBM, membrana glomerular basal). A terapia destes vascultídeos corresponde em grande parte aos princípios acima descritos para outros vascultídeos de pequenos vasos.
Doença de Behçet
A doença de Behçet caracteriza-se por um amplo espectro clínico e por um curso de recaída. A vasculite na doença de Behçet pode afectar os vasos arteriais e/ou venosos de todos os calibres. A vasculite das artérias pulmonares com formação consecutiva de aneurisma e ruptura no tecido pulmonar é a causa directa mais comum de morte. As complicações graves são também causadas por trombose venosa inflamatória, envolvimento cardíaco e cerebral.
A doença de Behçet tem algumas semelhanças clínicas impressionantes com a doença de Crohn, tais como a presença de colite, formações enterocolíticas e fístulas perianais, afetas mucocutâneas, artrites, eritema nodoso e uveíteos. A terapia das manifestações gastrointestinais na doença de Behçet reflecte isto: para além dos glicocorticóides, ácido 5-aminosalicílico (5-ASA), azatioprina e, se houver uma resposta insuficiente, são utilizados bloqueadores de TNFalpha. Dependendo da gravidade, esteróides tópicos, colchicina ou o inibidor de fosfodiesterase-4 apremilast (Otezla®), aprovado para este fim, podem ser utilizados para a terapia de aphthae mucocutaneous. No envolvimento ocular agudo, os glicocorticóides sistémicos (por exemplo 1g de metilprednisolona i.v. para a uveíte hypopyon) são recomendados principalmente, sempre em combinação com um DMARD, em primeiro lugar a ciclosporina A ou azatioprina, ou com interferon-alfa ou um bloqueador TNFalpha (infliximab ou adalimumab) [32]. Para o envolvimento vasculítico das artérias pulmonares, os glucocorticoides de alta dose em combinação com a ciclofosfamida ou bloqueadores de TNFalpha são eficazes. As intervenções não medicamentosas são também utilizadas para a doença grave de Behçet. Quando há uma ameaça de hemorragia de um grande aneurisma de artéria pulmonar, o tratamento de embolização é principalmente recomendado em vez da revisão de vasos cirúrgicos torácicos abertos. Em caso de hemorragia gastrointestinal grave, perfuração intestinal iminente ou estrangulamentos intestinais, os pacientes devem ser submetidos a uma cirurgia de emergência.
Mensagens Take-Home
- Em todos os vasculípedos, o primeiro objectivo do tratamento é alcançar uma remissão clínica tão completa quanto possível. Segue-se a fase de manutenção da remissão, que pode durar vários anos.
- O MTX nunca deve ser combinado com sulfometoxazol-trimetoprim no tratamento da RZA, caso contrário pode ocorrer uma mielossupressão severa.
- Um novo mecanismo de acção promissor para o tratamento da APG generalizada, bem como de outras AAV, é a inibição do sistema complemento.
- A vasculite sistémica de pequenos vasos em GPA pode afectar todos os órgãos. A infecção dos rins conduz tipicamente a uma glomerulonefrite “pauci-imune” (RPGN) progressiva e necrosante rápida, que pode levar a uma insuficiência renal grave se não for tratada.
Literatura:
- Jardel S, et al: Mortality in systemic necrotizing vasculitides: A retrospective analysis of the French Vasculitis Study Group registration. Autoimune Rev 2018 Jul; 17(7): 653-659.
- Jennette JC, Falk RJ, Bacon PA, et al: 2012 revista International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum 2013 Jan; 65(1): 1-11.
- Chung L, et al: Uso bem sucedido do Rituximab para a Vasculite Cutânea. Arch Dermatol 2006; 142(11): 1407-1410; doi:10.1001/archderm.142.11.1407.
- www.rheuma-net.ch/de/fachinformationen/behandlungsempfehlungen
- Villiger PM, Adler S, Kuchen S, et al: Tocilizumab para indução e manutenção de remissão em arterite de células gigantes: um ensaio fase 2, aleatório, duplo-cego, controlado por placebo. Lancet 2016; 387: 1921-1927.
- Stone JH, Tuckwell K, Dimonaco S et al: Ensaio de tocilizumab em arterite de células gigantes. N Engl J Med 2017; 377: 317-328.
- Christ L, et al: A Proof of Concept Study to Assess the Efficacy of Tocilizumab in Combination with Ultra-Short Glucocorticoid Administration to Treat Newly Diagnosed Giant Cell Arteritis – a 24 Week Analysis. Artrite Rheumatol 2020; 72 (suppl 10).
- Adler S, et al: Risco de recaída após descontinuação da terapia com tocilizumab em arterite de células gigantes. Reumatologia (Oxford) 2019 Set 1; 58(9): 1639-1643.
- Hellmich B, et al.: 2018 Actualização das recomendações da EULAR para a gestão da vasculite de grandes embarcações. Ann Rheum Dis 2020 Jan; 79(1): 19-30.
- Gadola SD, Gross WL: A renascença da inflamação granulomatosa em AAV. Nat Rev Rheumatol 2012 Jan 10; 8(2): 74-76.
- Holle JU, et al: As massas orbitais em granulomatose com poliangite estão associadas a um curso refractário e a uma elevada carga de danos locais. Reumatologia 2013; 52: 875882.
- DeRemee RA, et al: Granulomatose de Wegener: observações sobre o tratamento com agentes antimicrobianos. Case Reports Mayo Clin Proc 1985 Jan; 60(1): 27-32; doi: 10.1016/s0025-6196(12)65279-3.
- Stegeman CA, et al: Associação de porte nasal crónico de Staphylococcus aureus e taxas mais elevadas de recaídas na granulomatose de Wegener. Ann Intern Med 1994 1 de Janeiro; 120(1): 12-17.
- Rennie Rhee D, et al: Bactérias Nasais Associadas à Actividade de Doenças e Níveis de ANCA em Granulomatose com Poliangite. Artrite Rheumatol 2020; 72 (suppl 10).
- Taylor GB, et al: Uma revisão clinicopatológica de 34 casos de doença inflamatória da mama mostrando uma associação entre a infecção por córneas e mastite granulomatosa. Patologia 2003 Abril; 35(2): 109-119.
- Dobinson HC, et al: Opções de Tratamento Antimicrobiano para Mastite Granulomatosa Causada por Espécies de Corynebacterium. J Clin Microbiol 2015 Set; 53(9): 2895-2899.
- Holle JU, Dubrau C, Herlyn K, et al: Rituximab para granulomatose refractária com poliangite (granulomatose de Wegener): comparação da eficácia em manifestações granulomatosas versus vasculíticas. Ann Rheum Dis 2012; 71: 327-333.
- Voswinkel J, Mueller A, Kraemer JA, et al: B lymphocyte maturation in Wegener’s granulomatosis: a comparative analysis of VH genes from endonasal lesions, Ann Rheum Dis 2006; 65: 859-864.
- Voswinkel J, Assmann G, Held G et al: Análise monocelular de linfócitos B da granulomatose de Wegener: Os receptores de células B apresentam maturação por afinidade dentro das lesões granulomatosas. Clin Exp Immunol 2008; 154: 339-345.
- Knight A, et al: cancro urinário da bexiga na granulomatose de Wegener: riscos e relação com a ciclofosfamida. Ann Rheum Dis 2004.
- Thiel J, et al: Cinética de repovoamento celular B após tratamento rituximab em vasculites associadas ao ANCA em comparação com artrite reumatóide, e doenças do tecido conjuntivo: um estudo observacional longitudinal em 120 pacientes. Arthritis Res Ther 2017 18 de Maio; 19(1): 101.
- Merkel P, et al: The Effect on Renal Function of the Complement C5a Receptor Inhibitor Avacopan in ANCA-Associated Vasculitis. Artrite Rheumatol 2020; 72 (suppl 10).
- Huizenga N, et al: Tratamento da Vasculite Citoplasmática Anticorpo-Associada à Antineutrofílica Agressiva com Eculizumab. Kidney Int Rep 2020 Abr; 5(4): 542-545.
- Smtih R, et al: Acompanhamento Alargado de Pacientes Recrutados para um Ensaio Randomizado e Controlado de Rituximab contra Azathioprine após Indução de Remissão com Rituximab para Pacientes com Vasculite Associada a ANCA e Doença de Recaída. Artrite Rheumatol 2020; 72 (suppl 10).
- Guillevin L, et al: Rituximab versus azatioprina para manutenção na vasculite associada à ANCA. N Engl J Med 2014 6 de Novembro; 371(19): 1771-1780.
- Tzelepis GE, et al: Prevalência e resultado da fibrose pulmonar na poliangite microscópica. Revista Respiratória Europeia 2010; 36: 116-121.
- Sinico RA, et al: Envolvimento Renal na síndrome de Churg-Strauss. Am J Kidney Dis 2006 Maio; 47(5): 770-779.
- Wolf J, et al: Complicações neurológicas da síndrome de Churg-Strauss: um estudo monocêntrico prospectivo. Neurologia actual 2009; 36: V188.
- Conron M, Beynon HL: Síndrome de Churg-Strauss. Tórax 2000 Out; 55(10): 870-877.
- Solans R, et al: Síndrome de Churg-Strauss: resultado e acompanhamento a longo prazo de 32 pacientes. Reumatologia (Oxford) 2001; 40: 763-771.
- Braun G, et al: Vasculite crioglobulinaémica: classificação e aspectos clínicos e terapêuticos. Postgrad Med J 2007 Fev; 83(976): 87-94; doi: 10.1136/pgmj.2006.046078.
- Bettiol A, et al: Tratamento dos Diferentes Fenótipos da Síndrome de Behçet. Immunol frontal 2019 Dez 6; 10: 2830: doi: 10.3389/fimmu.2019.02830. eCollection 2019.
InFo DOR & GERIATURA 2020; 2(2): 12-19









