
Uma linfadenopatia persistente é uma indicação clara para diagnósticos histopatológicos adicionais. Uma história médica detalhada bem como o conhecimento sobre doenças anteriores, manipulações e terapias medicamentosas são essenciais para um diagnóstico definitivo do linfoma. Há casos que se encontram em zonas cinzentas em termos de classificação. A presença de rearranjos MYC é um indiscutível biomarcador de prognóstico em DLBCL, tal como a co-expressão fenotípica das proteínas myc e bcl2. A resposta à terapia rituximab está relacionada com a expressão do CD20. Os estudos moleculares estão a produzir novos parâmetros de previsão genética para uma resposta específica a terapias orientadas no linfoma.
Com uma quota relativa de 5%, os linfomas representam o quinto grupo mais frequente de doenças malignas em ambos os sexos. Especialmente os linfomas de células B maduras (anteriormente “linfomas não-Hodgkin”) mostram a mais forte incidência de malignoma em crescimento no mundo industrializado após o melanoma.
As razões para isto são desconhecidas, mas podem estar relacionadas com o aumento da esperança de vida, a crescente incidência de doenças auto-imunes e a associada utilização generalizada de (novos) imunossupressores, ou a crescente exposição a certos pesticidas e herbicidas. A incidência de linfoma é actualmente de cerca de 25 casos/100.000 habitantes/ano.
O progresso da terapia oncológica é claro no caso dos linfomas. As taxas de sobrevivência dos linfomas nodais mais comuns, tais como o linfoma difuso de grandes células B (DLBCL), o linfoma folicular (FL), o linfoma clássico de Hodgkin (HL) e o linfoma linfoblástico de células B (B-LBL, equivalente nodal da leucemia linfoblástica aguda de células B), são de cerca de 60% (DLBCL) e cerca de 80% (as restantes entidades). Isto explica a elevada prevalência do linfoma.
Definição
Os linfomas são definidos como doenças malignas, neoplásicas de linfócitos B, T ou NK imaturos ou maduros em órgãos do sistema linfático (nodal) ou fora desses órgãos (extranodal). Podem ser leucémicas ou sem exsudação (linfomas no sentido estreito).
Clínica
Linfomas nodais presentes com linfomas localizados ou generalizados, persistentes (mais de três semanas), frequentemente linfadenopatia progressiva com ou sem sintomas gerais (B) (febre, suores nocturnos, perda de peso), envolvimento de órgãos, alterações cutâneas (prurido, eritema) ou sinais de insuficiência de medula óssea (anemia, petéquias, tendência para a infecção).
Uma linfadenopatia persistente, especialmente acompanhada pelos sintomas mencionados, é portanto uma indicação clara para diagnósticos posteriores.
Diagnósticos
O diagnóstico baseado em tecidos é indispensável nos linfomas, uma vez que as alterações histopatológicas dos tecidos são as pedras angulares da dignidade e da determinação da entidade. Com base nestas alterações, é possível distinguir entre benigno e maligno e o grau de maturação dos linfócitos afectados por microscopia convencional de luz. Através de outros métodos microscópicos in situ, tais como a imuno-histoquímica (expressão de proteínas) ou a hibridação in situ por fluorescência (aberrações cromossómicas recorrentes), afiliação da linhagem (linhagem B, T ou NK), estádio exacto de desenvolvimento (por exemplo, célula B do centro germinal), expressões de marcadores patológicos (por exemplo, expressão de marcadores de células T em células B como na leucemia linfocítica crónica de células B) podem ser determinadas. [B-CLL]) ou translocação cromossómica existente (por exemplo, t[14;18] no FL) pode ser determinado e pode ser feito um diagnóstico exacto. Este é um pré-requisito básico para uma terapia oncológica específica. Em casos diagnósticos difíceis, o ADN pode ser obtido a partir do material (fixado em formol e incluído em parafina) e posteriormente analisado para a clonagem de células B e T, translocações e mutações pontuais.
Classificação
Os linfomas são classificados de acordo com a actual classificação da OMS. O princípio dominante é o diagnóstico integrador, que considera a inclusão da morfologia histopatológica do linfoma, os fenótipos (padrão de expressão das proteínas), os genótipos (aberrações cromossómicas recorrentes) e a clínica como sendo de igual importância na classificação das entidades.
A separação primária dos linfomas em linfomas de Hodgkin e linfomas não-Hodgkin foi abandonada. Devido à sua morfologia característica, apresentação clínica e excelente resposta a terapias específicas, HL continua a ser gerida como uma entidade distinta.
Novas categorias: Devido à complexidade biológica de certas doenças, duas das chamadas “categorias de zona cinzenta” foram introduzidas pela OMS:
- Linfomas não classificáveis de células B com características intermédias entre um DLBCL e um HL
- Linfomas não classificados de células B com características intermédias entre um linfoma DLBCL e um linfoma de Burkitt (BL).
Para além da sobreposição clínica e morfológica entre as entidades individuais em ambas as categorias, estudos moleculares mostram uma sobreposição significativa de genes expressos entre linfomas mediastinais primários de grandes células B e HL, por um lado, e entre DLBCL e BL individuais, por outro.
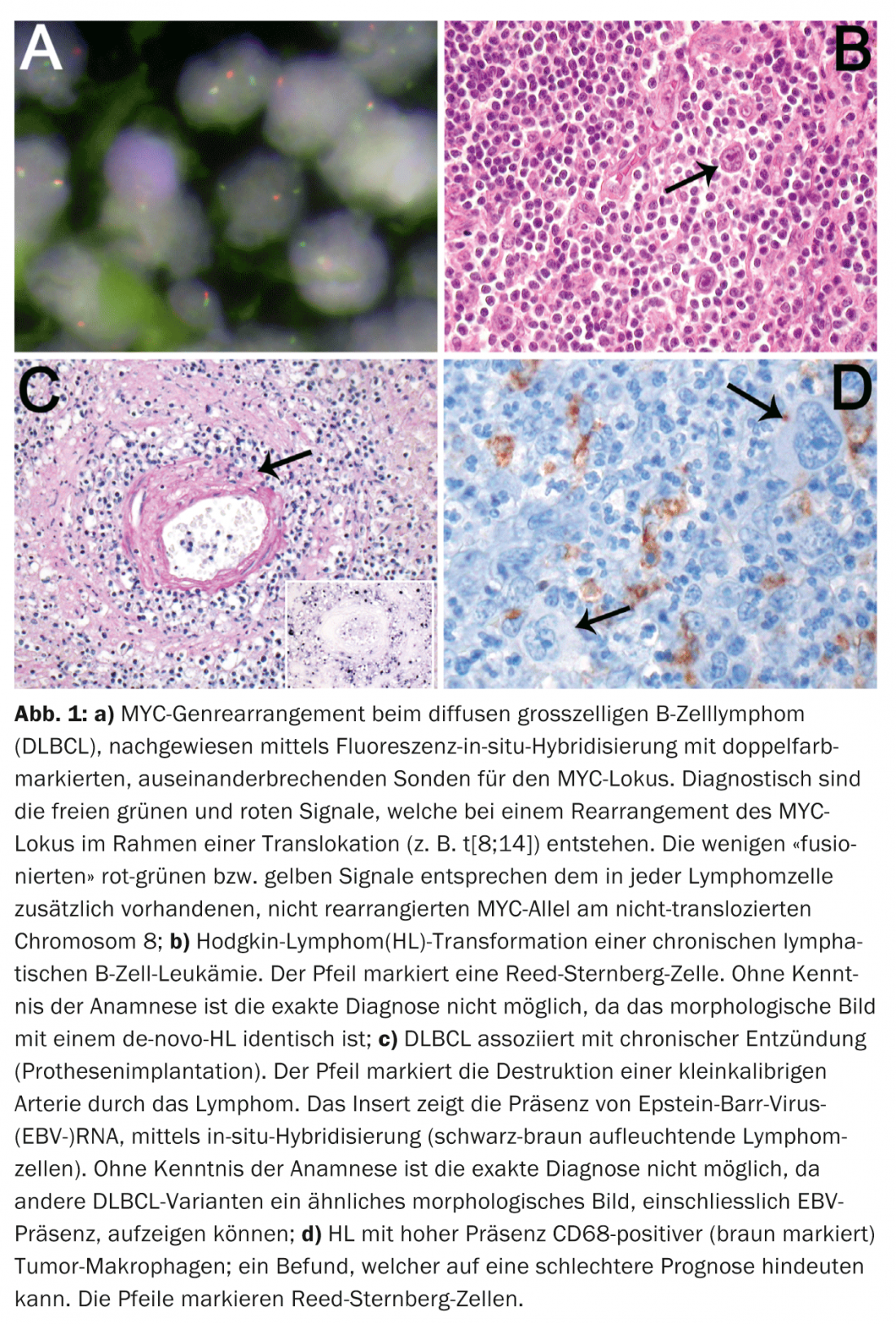
A evolução clínica desfavorável dos casos DLBCL altamente proliferativos com arranjo genético MYC (Fig. 1a) justifica ainda mais a introdução de uma categoria de zona cinzenta para dar seguimento a estes casos, que têm uma resposta inadequada às terapias DLBCL padrão, em ensaios terapêuticos prospectivos.
Categorias diagnósticas baseadas em parâmetros clínicos: Na classificação da OMS, o contexto relacionado com o doente, como a idade, a terapia medicamentosa anterior ou em curso, especialmente a terapia imunossupressora, e a localização foram fortemente considerados. Quatro entidades são definidas pela idade dos pacientes:
- FL pediátrico
- Linfoma pediátrico da zona marginal nodal B de células B (MZL)
- Vírus Epstein-Barr (EBV) – doença linfoproliferativa associada às células T da infância
- DLBCL associado à EBV nos idosos.
A justificação para a introdução destas entidades é a associação da emergência com a imaturidade ou senescência do sistema imunitário. Enquanto os três primeiros são claramente raros, a proporção relativa destes últimos é de 3% de todos os DLBCL. Embora os estudos do Extremo Oriente indiquem uma clara agressividade deste linfoma, os nossos dados da Europa Central mostram uma agressividade clínica particular apenas em casos individuais.
Reflectindo a frequência e heterogeneidade da DLBCL, foram definidas variantes clinicopatológicas adicionais na nova classificação. Para algumas destas variantes, tais como DLBCL associado a inflamação crónica (piroforax, osteomielite crónica, implantes de corpo estranho infectados, isto é, próteses vasculares/jointas, úlceras crónicas da pele, articulações artríticas na artrite reumatóide), o conhecimento da clínica é indispensável em termos de classificação. (Fig. 1b). Dado que 70% destes linfomas estão associados ao EBV e ocorrem principalmente em pacientes mais idosos, a diferenciação do DLBCL associado ao EBV nos idosos só é possível com base em evidências anamnésticas.
O conhecimento da clínica é também indispensável para o diagnóstico de doenças iatrogénicas, associadas à imunodeficiência, linfoproliferativas (terapia com imunossupressores), bem como para o diagnóstico de linfoproliferações pós-transplante (transplante de órgãos ou de medula óssea alogénica). O mesmo se aplica ao diagnóstico de linfomas secundários, transformados, “pequenos linfomas de células B” (B-CLL, MZL, FL), que se podem transformar num DLBCL, num linfoma não classificável de células B com características intermédias entre um DLBCL e um BL, bem como num HL. A indicação anamnéstica de um linfoma indolente anterior do grupo dos “pequenos linfomas de células B” é decisiva para a classificação correcta de tais lesões. Isto é significativo porque DLBCL ou HL (Fig. 1c), que se transformam a partir desses linfomas de células B, têm um curso significativamente mais agressivo do que os seus análogos de novo.
Prognóstico e previsão
Numerosos estudos ao longo da última década visaram estabelecer factores prognósticos significativos relacionados com tumores nos linfomas nodais mais comuns, tais como DLBCL, FL e HL. Contudo, os factores de prognóstico clínico conhecidos, tais como o Índice Prognóstico Internacional (IPI) em DLBCL, o Índice Prognóstico Internacional FL (FLIPI) e o Índice Prognóstico Internacional (IPS) em HL não foram ultrapassados pela inclusão de factores de prognóstico relacionados com tumores. Com excepção de algumas variantes de DLBCL que estão associadas a um prognóstico bastante pior, como a DLBCL rica em células T e histocitos, a granulomatose linfomatóide e a DLBCL intravascular, apenas a detecção de rearranjos MYC é um parâmetro prognóstico indiscutível relacionado com tumores em DLBCL. Três novos e independentes estudos de grande envergadura demonstraram o papel prognóstico desfavorável da co-expressão fenotípica das proteínas myc e bcl2 (o chamado “DLBCL fenotípico de duplo efeito”). Estudos em HL e FL mostraram um efeito prognóstico da composição das células T de fundo. Estudos recentes de expressão genética também demonstraram que as assinaturas de macrófagos associados a tumores podem influenciar significativamente a sobrevivência em HL, como reflectido morfologicamente por níveis elevados de macrófagos de tecido em doentes com prognóstico desfavorável (Fig. 1d).
Em resumo, os marcadores prognósticos no linfoma ainda não estão prontos para a prática diária. No entanto, a procura de parâmetros prognósticos associados ao microenvironmento tumoral parece promissora, especialmente porque este ambiente também poderia ser manipulado terapeuticamente sem medo de o tumor desenvolver resistência.
Outro campo de investigação diz respeito ao estabelecimento de marcadores preditivos, nomeadamente os biomarcadores que indicam a resposta ou não resposta a uma terapia. Embora os ensaios clínicos não tenham investigado especificamente a expressão do CD20 no estabelecimento do rituximab terapêutico anti-CD20, a experiência mostra que apenas os linfomas de células B que expressam o CD20 respondem a esta terapia. A determinação da expressão CD20 em linfomas é assim um exemplo de um marcador histopatológico determinável e preditivo.
Dados recentes sugerem que a sensibilidade específica aos inibidores de Bruton cinase (por exemplo ibrutinibe), agentes promotores de apoptose (por exemplo obatoclax) e inibidores de fosfoinosite 3-quinase (incluindo inibidores de mTOR como o everolimus) pode ser prevista a partir de alterações genéticas específicas nas células linfóides.
Prof. Alexandar Tzankov, MD
Prof. Dr. med. Stephan Dirnhofer
Literatura:
- Roman E, Smith AG: Histopatologia 2011; 58: 4-14.
- Swerdlow SH, et al: Classificação da OMS de tumores de tecidos hematopoiéticos e linfóides. Lyon: IARC; 2008.
- Hoeller S, et al: Hum Pathol 2010; 41: 352-357.
- Tzankov A, et al.: Mod Pathol 2013; doi: 10.1038/modpathol.2013
- Hu S, et al: Blood 2013; 121: 4021-4031.
- Steidl C, et al: N Engl J Med 2010; 362: 875-885.
- Tzankov A, et al: Haematologica 2008; 93: 193-200.
- Rahal R, et al: Nat Med 2014; 20: 87-92.
- Wenzel SS, et al: Leucemia 2013; 27: 1381-1390.
- Pfeifer M, et al: Proc Natl Acad Sciad U S A. 2013; 110: 12420-12425.
InFo Oncologia & Hematologia 2014; 2(2): 5-7









