
A imunofluorescência direta e indireta continua a ser o padrão de ouro para a deteção de anticorpos específicos e auto-anticorpos no penfigoide bolhoso. Os esteróides tópicos altamente eficazes ou os corticosteróides sistémicos são recomendados como terapêutica de primeira linha. Em caso de ausência de resposta ou de contra-indicações, podem ser utilizados medicamentos imunomoduladores e imunossupressores. De acordo com vários relatos de casos, o omalizumab e o dupilumab provaram ser opções de tratamento eficazes e estão atualmente a ser investigados outros produtos biológicos.
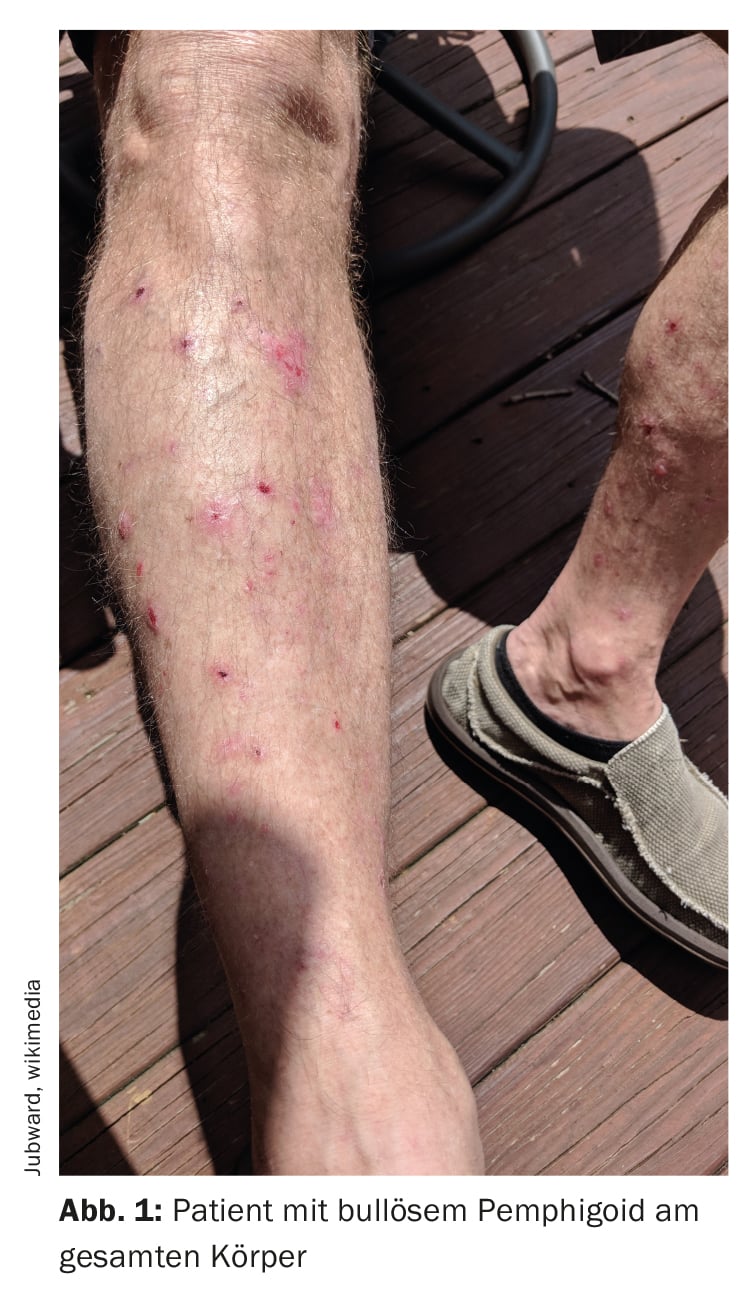
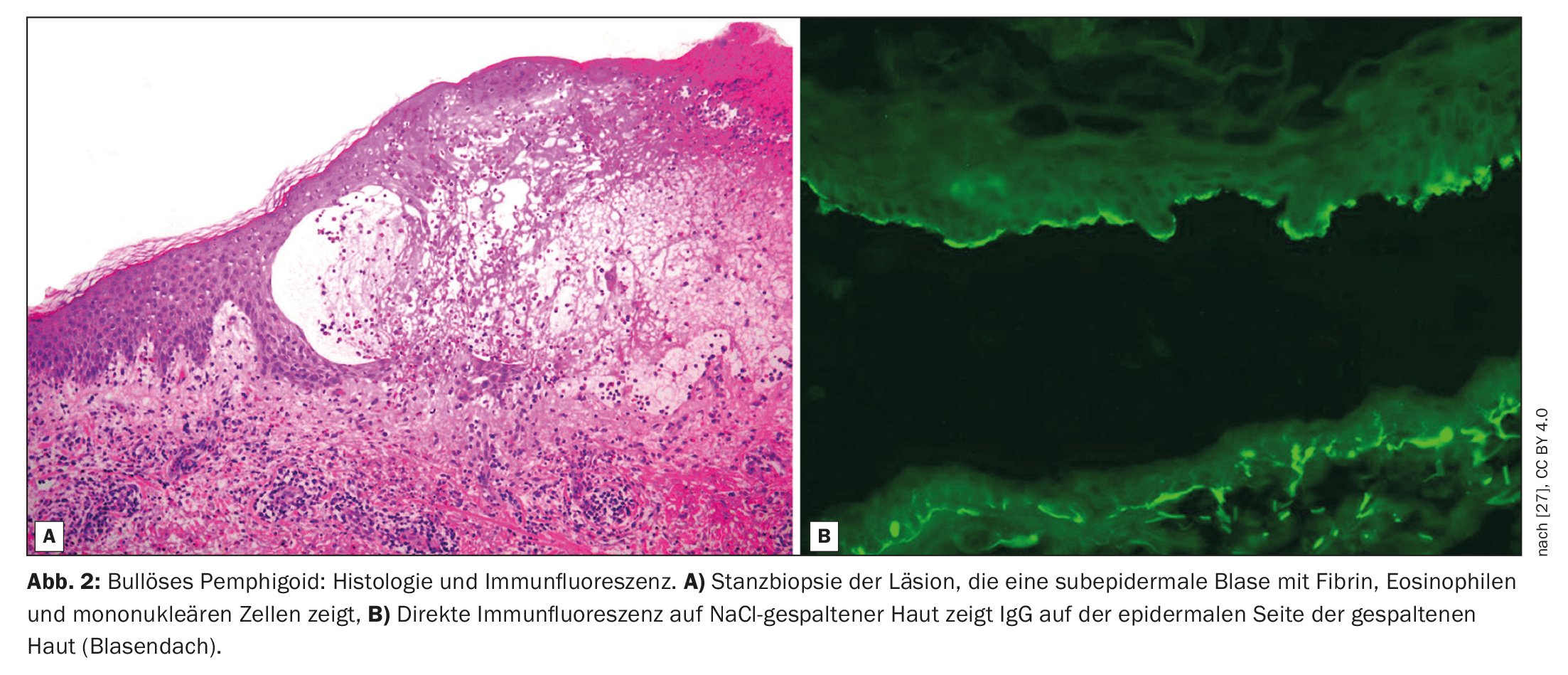
O penfigoide bolhoso (PB) caracteriza-se pela formação de bolhas subepidérmicas, ou seja, bolhas na zona de junção dermoepidérmica (Fig. 1, Fig. 2) [1]. É a dermatose autoimune bolhosa mais comum na Suíça e na Europa, segundo o Prof. Dr. Luca Borradori, Diretor Clínico e Médico Chefe da Clínica Universitária de Dermatologia, Inselspital Bern [2]. Nas últimas décadas, tem-se observado também um aumento da incidência de BP neste país. As possíveis causas para este facto incluem o envelhecimento da população, a associação com doenças neurológicas cada vez mais comuns e determinados medicamentos, bem como uma maior sensibilização para as variantes atípicas sem formação de bolhas [28]. O tratamento da PA tem como objetivo controlar a doença, aliviar os sintomas de prurido e melhorar a qualidade de vida das pessoas afectadas. Os métodos de tratamento dependem essencialmente da gravidade da doença. Deve ser prestada atenção a um perfil de risco-benefício favorável do respetivo tratamento, diz o Prof. Borradori.
Suspeita clínica de BP: a serologia dá-lhe a certeza
O diagnóstico da PB baseia-se numa combinação de critérios, incluindo características clínicas, achados de microscopia ótica e deteção de auto-anticorpos na pele por imunofluorescência direta (IFD) [3]. A manifestação clínica clássica da PA é uma erupção cutânea muito pruriginosa com bolhas generalizadas (bolhas salientes e pruriginosas de vários tamanhos cheias de líquido seroso). Nas fases iniciais ou nas variantes atípicas, no entanto, apenas podem estar presentes lesões escoriadas, eczematosas ou urticariformes, localizadas ou generalizadas [4]. As membranas mucosas são afectadas por 10-30%. O achado típico na imunofluorescência direta é a deteção de imunoglobulina linear (Ig)G ligada à zona de junção dermo-epidérmica, mais raramente também IgM e IgA. Estes auto-anticorpos ligam-se principalmente às proteínas BP180 e BP230, que são componentes dos hemidesmossomas. Estas representam uma importante ligação celular e têm uma função essencial para a estabilidade mecânica da pele [1].
A fim de confirmar o diagnóstico suspeito de PA, as seguintes etapas adicionais de clarificação do diagnóstico laboratorial são recomendadas nas orientações s2K de um grupo de peritos da EADV, actualizadas em 2022 [3]:
- Deteção de auto-anticorpos IgG circulantes contra a zona da membrana basal (BMZ) por microscopia de imunofluorescência indireta (IIF) utilizando pele humana normal clivada com solução de NaCl.
- Deteção de auto-anticorpos IgG anti-BP180-NC16A e/ou de auto-anticorpos IgG anti-BP230 por ELISA (ensaio de imunoabsorção enzimática).
- Os novos testes “multivariados” são também recomendados para a deteção de auto-anticorpos IgG anti-BMZ circulantes. Estes ensaios BIOCHIP mosaico combinam diferentes substratos antigénicos. Nos raros casos de PA em que os anticorpos circulantes anti-BMZ não são detectáveis por microscopia de imunofluorescência indireta ou por ELISA comercialmente disponível, recomenda-se a realização de testes adicionais para aumentar a sensibilidade do diagnóstico e excluir outras doenças auto-imunes da junção dermo-epidérmica (em particular, penfigoide anti-P200 ou epidermólise bolhosa adquirida). Pode encontrar uma panorâmica dos testes correspondentes nas directrizes s2K publicadas no JEADV [3].
Fisiopatologia: estão também envolvidos mecanismos independentes do complemento
As relações fisiopatológicas da PA são muito complexas [4]. Por um lado, a ativação do sistema do complemento com a produção de anafilatoxinas e a ativação da resposta imune inata com o subsequente recrutamento e ativação de basófilos, eosinófilos, neutrófilos, monócitos/macrófagos e mastócitos desempenham um papel fundamental na PA [5,6, 28-31]. Por outro lado, nos últimos anos, os investigadores têm vindo a descobrir cada vez mais mecanismos inflamatórios independentes do complemento relacionados com os anticorpos da PA. Os queratinócitos, em particular, parecem ser capazes de segregar um grande número de citocinas pró-inflamatórias relevantes do ponto de vista patogénico [4,7,32].
Um estudo mostrou que tanto o BP-IgG como o BP-IgE são capazes de se ligar diretamente à superfície de queratinócitos humanos em cultura, com subsequente perda de hemidesmossomas na zona da membrana basal [8]. Os eosinófilos estão envolvidos na patogénese da PA, mediando os efeitos dos anticorpos IgE anti-BP180 e contribuindo para a separação dermo-epidérmica. Na presença de IgG ou IgE, os eosinófilos podem mediar diretamente a separação dermo-epidérmica [9,10]. Freire et al. encontraram níveis elevados de IgE anti-BP180 e anti-BP230 utilizando ELISA e imunofluorescência em comparação com controlos saudáveis [11].
Os auto-anticorpos IgE anti-BP180 estão presentes na maioria dos doentes com PA e os seus níveis estão correlacionados com a atividade da doença [12–15]. Além disso, os estudos de imunofluorescência direta mostraram que a maioria dos doentes com PB tinha células IgE+ na pele, em contraste com os controlos saudáveis. De um modo geral, as provas sugerem que existe uma via adicional independente do complemento, dependente de Th2 e mediada por eosinófilos que contribui para a lesão dos tecidos e para as características clínicas da BP [4].
| “Tratar o alvo” com monitorização e acompanhamento do progresso |
| – O grupo de peritos da EADV recomenda geralmente uma duração de tratamento entre 9 e 12 meses para a BP [3]. |
| – Recomenda-se a interrupção do tratamento em doentes que tenham estado sem sintomas durante pelo menos 1-6 meses com uma terapêutica mínima com prednisona oral (0,1 mg/kg/dia), propionato de clobetasol (10 g/semana) ou imunossupressores. A interrupção do tratamento é igualmente influenciada pelo estado geral do doente e pela presença de determinadas comorbilidades. |
| – anti-BP180 ELISA (ou seja, >27 U/mL, conforme determinado pelo teste MBL**) e, em menor grau, estudos DIF$ foram relatados como tendo valor preditivo para a ocorrência de recaída após a interrupção do tratamento [24]. Pode valer a pena considerar a utilização destes testes antes de interromper o tratamento. |
| – Além disso, deve prestar atenção a uma possível insuficiência adrenal causada pela utilização de corticosteróides exógenos (CS) (também após a utilização de CS tópicos). |
| Recomenda-se um exame de acompanhamento no terceiro mês após a interrupção do tratamento. Este período parece ser suficiente para reconhecer a maioria das recaídas de PA [24–26]. Independentemente disso, a recorrência de comichão, escoriações e/ou lesões inflamatórias da pele deve ser sempre esclarecida por um médico. |
| ** MBL= Laboratórios Médicos e Biológicos $ DIF=Imunofluorescência direta |
Novas opções de tratamento: Os produtos biológicos como terapia alternativa
O quadro 1 [3] apresenta uma panorâmica das opções de tratamento atualmente disponíveis. As directrizes continuam a recomendar a utilização de corticosteróides tópicos superpotentes (TCS) ou esteróides sistémicos (prednisolona 0,5 mg/kg/d) como terapêutica de primeira linha. Em muitos casos, os sintomas podem ser contidos desta forma, mas “é preciso ter paciência”, explicou o Prof. Borradori [2]. A duração estimada do tratamento é de 9 a 12 meses. No entanto, há também doentes para os quais a terapêutica padrão não é eficaz ou está contra-indicada. “Precisamos de terapias boas, novas e eficazes”, sublinhou o orador [2]. A observação de que os doentes com PA têm frequentemente níveis séricos elevados de IgE e autoanticorpos IgE específicos para BP180 e BP230 em circulação apoia a ideia de que a IgE desempenha um papel na patogénese da PA [16,17]. A utilização de omalizumab, um anticorpo monoclonal humanizado que inibe a ligação da IgE ao seu recetor de alta afinidade (FcεRI), é, por conseguinte, uma escolha óbvia. Relatos de casos mostram que a terapêutica com omalizumab leva a uma forte diminuição da expressão de FcεRI nos basófilos circulantes e a uma forte redução das células FcεRI + na pele dos doentes tratados [16].
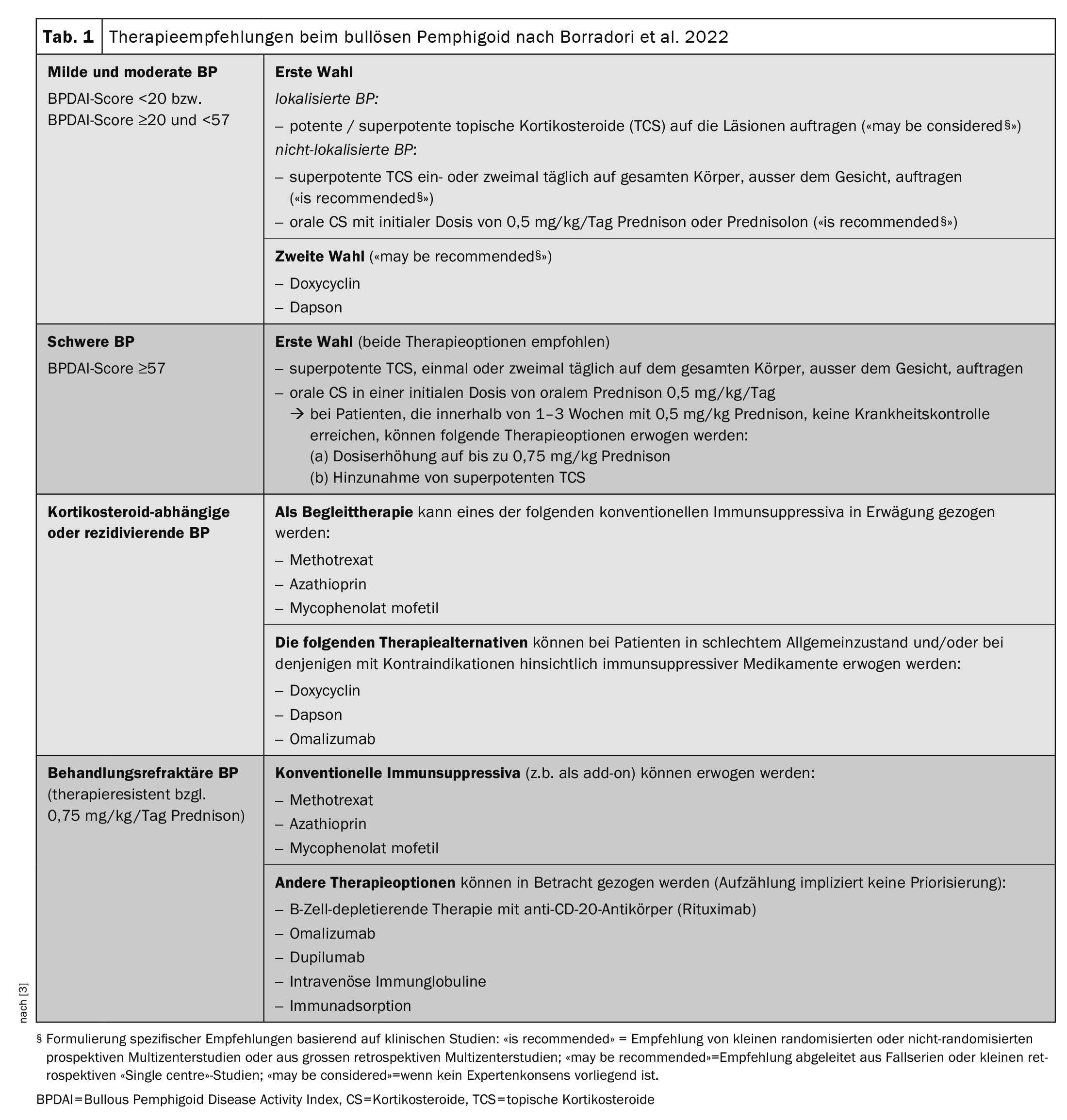
Outras terapias biológicas direccionadas que se revelaram promissoras na BP incluem o dupilumab, um anticorpo monoclonal IgG4 humano que se liga à subunidade α do recetor de IL-4, inibindo assim as vias de sinalização IL-4/IL-13 [18–21]. “Temos muitas séries de casos que mostram que o dupilumab tem um bom efeito em doentes com penfigoide bolhoso”, referiu o Prof. Borradori [19,22]. O medicamento biológico provou ser eficaz tanto em monoterapia como em combinação com uma terapia padrão. Na sequência de testes clínicos bem sucedidos na Fase II, a eficácia e a segurança do dupilumab na PA estão atualmente a ser investigadas no ensaio de Fase III LIBERTY-BP (NCT04206553) [23].
Congresso: Formação avançada conjunta BE-BS-ZH
Literatura:
- Diagnóstico e tratamento do pênfigo vulgar/foliáceo e do penfigoide bolhoso, AWMF guideline S2k 2019, Sociedade Dermatológica Alemã (DDG), registo AWMF n.º: 013-071.
- “Dr. Luca Borradori, Formação contínua conjunta das clínicas dermatológicas de Berna, Basileia, Zurique, Inselspital Bern, 25.05.2023.
- Borradori L, et al: Directrizes S2 K actualizadas para a gestão do penfigoide bolhoso iniciadas pela Academia Europeia de Dermatologia e Venereologia (EADV). J Eur Acad Dermatol Venereol 2022; 36(10): 1689-1704.
- Cole C, et al: Perspectivas sobre a patogénese do penfigoide bolhoso: O papel dos mecanismos independentes do complemento. Compass Dermatol 2022; 10 (4): 171-180.
- Liu Z, et al: A formação de bolhas subepidérmicas induzida por autoanticorpos humanos para BP180 requer intervenientes imunes inatos num modelo de ratinho com penfigoide bolhoso humanizado. J Autoimmun 2008; 31(4): 331-338.
- Liu Z, et al: Sinergia entre uma cascata de plasminogénio e a MMP-9 na doença autoimune. J Clin Invest 2005; 115(4): 879-887.
- Bao L, et al: A reatividade específica da subunidade de autoanticorpos contra a laminina-332 revela mecanismos inflamatórios directos nos queratinócitos. Front Immunol 2021; 12: 775412.
- Messingham KN, et al: Efeitos independentes de FcR dos autoanticorpos IgE e IgG no penfigoide bolhoso. J I 2011; 187(1): 553-560.
- Amber KT, et al: The Role of Eosinophils in Bullous Pemphigoid: A Developing Model of Eosinophil Pathogenicity in Mucocutaneous Disease (O Papel dos Eosinófilos no Penfigoide Bolhoso: Um Modelo em Desenvolvimento da Patogenicidade dos Eosinófilos na Doença Mucocutânea). Front Med (Lausanne). 2018; 5: 201.
- de Graauw E, et al: Evidência de um papel dos eosinófilos na formação de bolhas no penfigoide bolhoso. Allergy 2017; 72(7): 1105-1113.
- Freire PC, Munoz CH, Stingl G: Autoreactividade da IgE no Penfigoide Bolhoso: Eosinófilos e Mastócitos como Alvos Principais de Reactores Imunitários Patogénicos. Br J Dermatol 2017; 177(6): 1644-1653.
- Hashimoto T, et al: Deteção de autoanticorpos IgE para BP180 e BP230 e sua relação com características clínicas no penfigoide bolhoso. Br J Dermatol 2017; 177(1): 141-151.
- van Beek N, et al: Correlação dos níveis séricos de autoanticorpos IgE contra BP180 com a atividade da doença do penfigoide bolhoso. JAMA Dermatol 2017; 153(1): 30-38.
- Bing L, et al: Os níveis de IgE anti-BP180 NC16A não estão correlacionados com a gravidade da doença nas fases iniciais do penfigoide bolhoso. Arch Dermatol Res 2015; 307(9): 849-854.
- Salz M, et al: Níveis séricos elevados de IL-31 em doentes com penfigoide bolhoso correlacionam-se com números de eosinófilos e estão associados a BP180-IgE. J Dermatol Sci 2017; 87(3): 309-311.
- Seyed Jafari SM, et al: Efeitos do omalizumab na expressão de FcεRI e IgE na pele lesional do penfigoide bolhoso. Front Immunol 2019; 10: 1919.
- Messingham KN, Crowe TP, Fairley JA: A intersecção de auto-anticorpos IgE e eosinofilia na patogénese do penfigoide bolhoso. Front Immunol. 2019;10: 2331.
- Abdat R, et al: Dupilumab como uma nova terapia para o penfigoide bolhoso: uma série de casos multicêntricos. J Am Acad Dermatol 2020; 83(1): 46-52.
- Zhang Y, et al: Eficácia e segurança do dupilumab no penfigoide bolhoso moderado a grave. Front Immunol 2021; 12: 738907.
- Seyed Jafari SM, et al: Relato de caso: Combinação de Omalizumab e Dupilumab para penfigoide bolhoso recalcitrante. Front Immunol 2020; 11: 611549.
- Kaye A, et al: Dupilumab para o tratamento do penfigoide bolhoso recalcitrante. JAMA Dermatol 2018; 154(10): 1225-1256.
- Klepper EM, Robinson HN: Dupilumab para o tratamento do penfigoide bolhoso induzido por nivolumab: relato de um caso e revisão da literatura. Dermatol Online J. 2021;27(9): 1-6.
- Biblioteca Nacional de Medicina dos EUA. Um estudo para avaliar a eficácia e a segurança do dupilumab em doentes adultos com penfigoide bolhoso (LIBERTY-BP). Bethesda, MD: Biblioteca Nacional de Medicina dos EUA (2019).
- Bernard P, et al: Factores de risco de recaída em doentes com penfigoide bolhoso em remissão clínica: um estudo de coorte multicêntrico e prospetivo. Arch Dermatol 2009; 145: 537-542.
- Joly P, et al: Comparação de dois regimes de corticosteróides tópicos no tratamento de doentes com penfigoide bolhoso: um estudo multicêntrico aleatório. J Invest Dermatol 2009; 129: 1681-1687.
- Fichel F, et al: Factores clínicos e imunológicos associados à recidiva do penfigoide bolhoso durante o primeiro ano de tratamento: um estudo multicêntrico e prospetivo. JAMA Dermatol 2014; 150: 25-33.
- Michelerio A, Tomasini C: Bolhas e milia à volta do cateter de diálise peritoneal: Um caso de penfigoide bolhoso localizado. Dermatopathology 2022; 9(3): 282-286. www. mdpi.com/2296-3529/9/3/33,(último acesso em 10/08/2023).
- Holtsche MM, Boch K, Schmidt E: J Dtsch Dermatol Ges 2023; 21(4): 405-413.
- Chen R, et al: J Clin Invest 2001; 108(8): 1151-1158.
- Nelson KC, et al: J Clin Invest 2006; 116(11): 2892-900.
- Leighty L, et al: Arch Dermatol Res 2007; 299(9): 417-422.
- Schmidt E, et al: J Invest Dermatol 2000; 115(5): 842-848.
DERMATOLOGIE PRAXIS 2023; 33(5): 50-52 (publicado em 30.10.23, antes da impressão)









