
A detecção precoce, a estratificação do risco e uma terapia adaptada à esclerose sistémica (SSc) são de grande importância para poder contrariar atempadamente as limitações relacionadas com a doença e as manifestações potencialmente ameaçadoras da vida. A doença pulmonar intersticial (DPI) é uma manifestação orgânica de SSc com mortalidade significativa. Para além dos imunossupressores, o medicamento antifibrótico nintedanib também está disponível na Suíça há algum tempo e o tocilizumab foi recentemente aprovado nos EUA com base em resultados positivos da fase III.
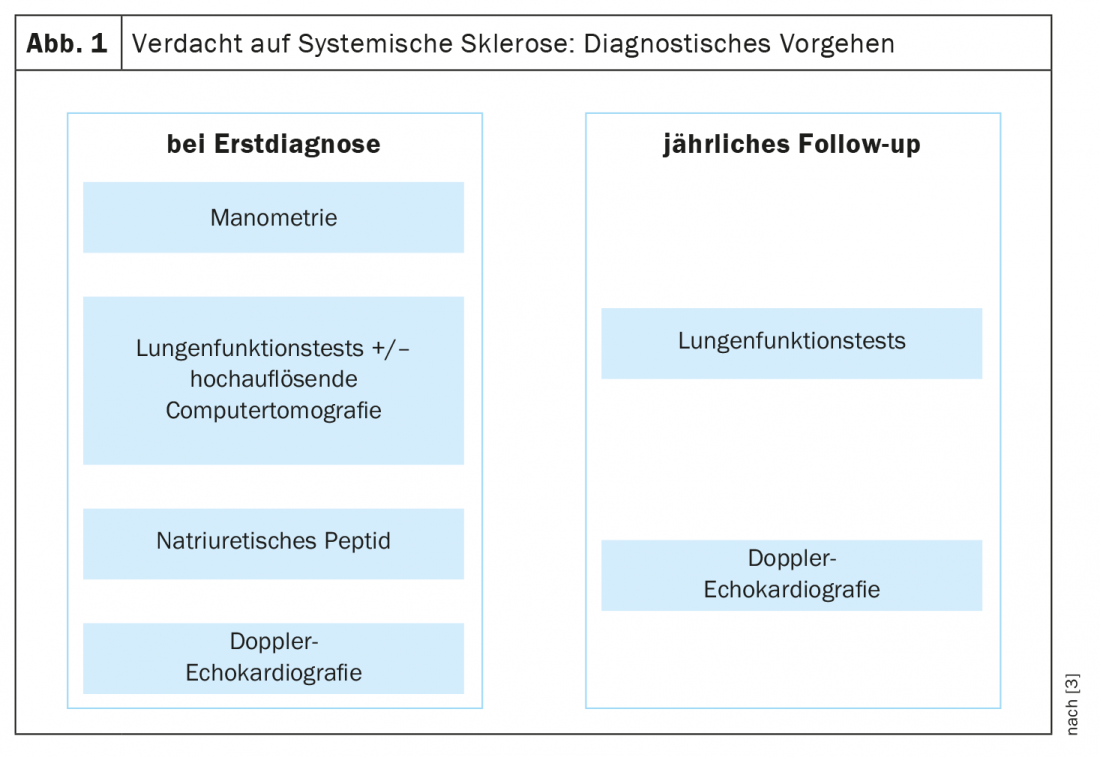
Para o diagnóstico precoce da esclerose sistémica, os critérios VEDOSS (“Very Early Diagnosis of Systemic Sclerosis”) foram estabelecidos há alguns anos atrás [1,2]. A síndrome de Raynaud, o inchaço dos dedos (“dedos inchados”), alterações nos capilares das pregas e a detecção de auto-anticorpos antinucleares (ANA) são preditivos de esclerose sistémica (SSc), explicou a Dra. Hanna Grasshoff, Clínica de Reumatologia e Imunologia Clínica, Hospital Universitário Schleswig-Holstein, Lübeck [1]. Os critérios de classificação estabelecidos em 2013 pelo American College of Rheumatology (ACR) e pela European League Against Rheumatism (EULAR) continuam a ser válidos [1,16]. Os ANA associados ao SSc mais comuns incluem o anti-centrómero-Ak (ACA) e o anti-topoisomerase-Ak (ATA, Scl70). As actuais descobertas empíricas confirmam que os critérios VEDOSS são adequados para a estratificação dos riscos dos pacientes. Isto também foi demonstrado num estudo publicado em 2021 no European Journal of Internal Medicine [3]. O orador resume o procedimento de diagnóstico da seguinte forma (Fig. 1) : “Os pacientes são submetidos a manometria esofágica, testes de função pulmonar e, se necessário, tomografia computorizada de alta resolução, se existirem anomalias, bem como peptídeo natriurético e ecocardiografia Doppler” [1]. Um ECG adicional pode ser útil [1]. Testes anuais de função pulmonar e ecocardiografias Doppler são recomendados durante o curso. Relativamente à hipertensão arterial pulmonar, incluindo SSc-PAH, foi publicada em 2022 uma nova directriz ESC/ERS [17]. Os valores alvo anteriores foram modificados e são agora os seguintes: mPAP (pressão média da artéria pulmonar) >20 mmHg, PAWP (pressão da cunha arterial pulmonar) ≤15 mmHg, PVR (resistência vascular pulmonar) ≥ 2 WU (unidades de madeira*).
* unidade tradicional de medição das resistências dos vasos
“Usar “Janelas de oportunidade
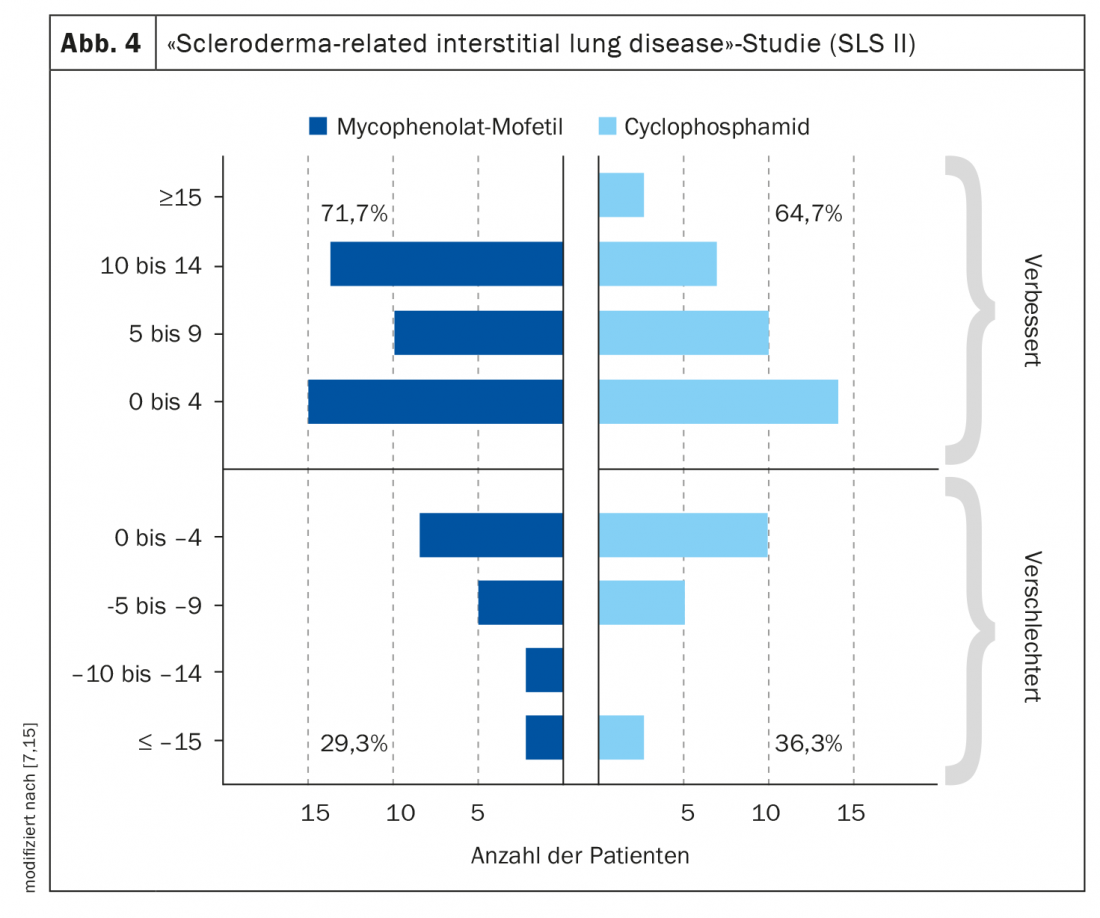
A distinção entre SSc cutâneo limitado e SSc cutâneo difuso ainda é relevante. De acordo com as recomendações da EULAR, o procedimento terapêutico depende do envolvimento dos órgãos [5]: 1. síndrome de Raynaud, 2. úlceras digitais, 3. SSc-PAH, 4. manifestação da pele e dos pulmões, 5. crise renal, 6. Manifestação gastrointestinal. Os dois medicamentos mais utilizados são a ciclofosfamida e o micofenolato mofetil (MMF), com base em dois ensaios controlados aleatorizados com resultados de eficácia semelhantes [6,7]. No Scleroderma Lung Study (SLS) II, a ciclofosfamida e o MMF alcançaram uma eficácia comparável, mas o MMF tinha o melhor perfil de segurança e tolerabilidade a longo prazo (Fig. 4). Portanto, o MMF é utilizado com mais frequência na prática clínica. De grande interesse, claro, é a questão das implicações que um diagnóstico precoce tem para a terapia. Num estudo de coorte retrospectivo publicado em 2022, os pacientes com SSc que manifestaram doença cutânea difusa ou doença pulmonar intersticial dentro de 6 anos após o início da doença foram divididos em grupos de intervenção precoce e retardada com base na duração da doença. No primeiro, o tratamento foi iniciado dentro de ≤18 meses desde o início da doença, no segundo apenas >18 meses após [4]. As opções de terapia medicamentosa utilizadas foram a ciclofosfamida, o MMF, o metotrexato ou o tocilizumabe. No grupo de intervenção precoce, a doença activa diminuiu significativamente de 79% para 42% (p=0,007), enquanto a alteração no grupo de intervenção tardia não foi estatisticamente significativa (68% para 42%; p=0,11). Globalmente, os resultados deste estudo apoiam a suposição de que existe uma “janela de oportunidade” para opções terapêuticas em pacientes com SSc.
SSc-ILD – espectro terapêutico alargado para complicação com risco de vida
Para manifestações cutâneas e pulmonares, a EULAR recomenda as seguintes opções de medicamentos: Metotrexato, ciclofosfamida, transplante autólogo de células estaminais hematopoiéticas, possivelmente MMF, possivelmente azatioprina. A doença pulmonar intersticial (DPI) na esclerose sistémica (SSc-ILD) é actualmente a causa de morte associada à doença mais comum em doentes com SSc [8]. A prevalência da ILD como complicação da SSc é de cerca de 50% numa coorte recente de base populacional [9]. A principal terapia medicamentosa utilizada até agora tem sido os imunossupressores, sendo os MMF o medicamento mais utilizado internacionalmente [10,11]. Estudos recentes sugerem que o nintedanib e o tocilizumab podem ajudar a retardar a deterioração da função pulmonar na SSc-ILD [12]. O Nintedanibe é um inibidor antifibrótico da tirosina quinase utilizado na doença intersticial do pulmão para prevenir a cicatrização do tecido pulmonar. Na Suíça, o nintedanib (Ofev®) foi aprovado desde 2020 para o tratamento da doença pulmonar intersticial associada à esclerose sistémica [12]. A dose recomendada é de 150 mg duas vezes por dia, com intervalos de cerca de 12 horas. Tocilizumab foi aprovado pela FDA dos EUA para o tratamento de SSc-ILD com base em dados de um ensaio da fase III [13].
A medicina de precisão aponta o caminho para o futuro
Um artigo de posição sobre transplante autólogo de células estaminais hematopoiéticas (ahSCT) para esclerose sistémica foi publicado pelo Grupo de Trabalho de Terapia com Células Estaminais da Sociedade Alemã de Reumatologia [14]. Consequentemente, ahSZT é razoável nas seguintes condições: duração máxima da doença de 4 anos, mRSS de min. 15, envolvimento de órgãos internos ou outros factores prognósticos desfavoráveis, resposta insuficiente à ciclofosfamida ou ao MMF. A SSc é uma doença muito heterogénea. “Precisamos de estratificação molecular a diferentes níveis OMICS a longo prazo”, diz a Dra. Grasshoff [1]. É aqui que entra a medicina de precisão. “O objectivo seria tratar a vasculopatia vasoactivamente, a inflamação imunomoduladora e a fibrose antifibrótica”, disse ela [1]. Os estudos que correlacionam a assinatura genética intrínseca com a resposta a diferentes medicamentos são uma abordagem promissora para uma estratégia de tratamento específica para um subgrupo com um perfil de risco-benefício optimizado.
Congresso: Congresso Alemão de Reumatologia
Literatura:
- “Esclerose sistémica – uma doença heterogénea”, Dra. Hanna Grasshoff, Congresso Alemão de Reumatologia, 31.08.-03.09.2022.
- Minier T, et al: Ann Rheum Dis 2014; 73(12): 2087-2093.
- Gonzalez Garcia A, Callejas-Rubio JL: Jornal Europeu de Medicina Interna 97(Suppl 113); DOI:10.1016/j.ejim.2021.12.012
- Yomono K, Kuwana M: Rheumatology (Oxford) 2022; 61(9): 3677-3685.
- Kowal-Bielecka O, et al: Ann Rheum Dis 2017; 76(8): 1327-1339.
- Tashkin DP, et al: NEJM 2006 (354): 2655-2666.
- Tashkin DP, et al: Lancet Respir Med 2016 (4): 708-719.
- Elhai M, et al: Ann Rheum Dis 2019; 78: 979-987.
- Hoffmann-Vold AM, et al: Am J Respir Crit Care Med 2019; 200: 1258-1266.
- Fernández-Codina A, et al: Arthritis Rheumatol 2018; 70: 1820-1828.
- Khanna D, et al: [abstract]. Artrite Rheumatol 2018, 70 (suppl 9). https://acrabstracts.org/
- Informação sobre drogas, www.swissmedicinfo.ch, (último acesso 07.11.2022)
- Khanna D, et al: Lancet Respir Med 2020; 8: 963-974
- Alexander T, Burmester G: Journal of Rheumatology. Edição 5/2020.
- Schneider U, et al: Z Rheumatol 2021; 80: 868-878.
- van den Hoogen F, et al: Arthritis Rheum 2013; 65(11): 2737-2347.
- Humbert M, et al: Grupo de Documentos Científicos ESC/ERS. EHJ 2022; 43(38): 3618-3731.
PRÁTICA DO GP 2022; 17(11): 16-17









