A terapia genética, a esperança de longa data da investigação médica, está finalmente a encontrar o seu caminho na clínica para curar doenças monogenéticas. Assim, a medicina tem à sua disposição uma nova terapia que pode tratar as doenças hereditárias não só de forma sintomática mas também causal. Outra abordagem de terapia genética é o tratamento de alterações genéticas adquiridas em tumores malignos usando células T CAR (receptor de antígeno quimérico).
A terapia genética, a esperança de longa data da investigação médica, está finalmente a encontrar o seu caminho na clínica para curar doenças monogenéticas. Assim, a medicina tem à sua disposição uma nova terapia que pode tratar as doenças hereditárias não só de forma sintomática mas também causal. Outra abordagem de terapia genética é o tratamento de alterações genéticas adquiridas em tumores malignos usando células T CAR (receptor de antígeno quimérico). Segue-se uma visão geral dos princípios biológicos moleculares e da aplicação clínica dos actuais procedimentos de terapia genética e celular.
Genes como plantas de todas as proteínas – mutações como causa de doenças monogénicas
Cada célula humana contém uma planta genética dentro do núcleo celular sob a forma de ácido desoxirribonucleico (ADN). O ADN é constituído por quatro blocos básicos de construção, as bases adenina (A), timina (T), guanina (G) e citosina (C), que, alinhadas em longas cadeias lineares, formam a estrutura básica dos cromossomas de uma célula humana. O ADN de cada célula contém unidades codificadoras individuais, os genes. Cada gene consiste numa região codificadora e numa unidade reguladora, o promotor, cuja actividade pode ser amplificada por um elemento potenciador, se necessário. Cada gene codifica a estrutura de uma proteína através da sequência dos quatro blocos básicos de construção do ADN. As proteínas são compostas por 20 blocos de construção básicos, os aminoácidos, que se dobram em uma ou mais cadeias lineares para formar proteínas. Apenas proteínas devidamente dobradas podem desempenhar a sua função biológica na célula.
Doenças monogenéticas
Se ocorrer uma reparação defeituosa após danos no ADN, a sequência dos blocos básicos de construção do ADN pode ser alterada. Alternativamente, os blocos básicos de construção de ADN também podem ser perdidos. Estas alterações são chamadas “mutações”. Se ocorrer uma mutação numa secção do ADN que codifica uma proteína, isto pode alterar a estrutura da proteína aí codificada: A proteína defeituosa sintetizada de acordo com o plano do ADN não pode cumprir a sua função natural na célula ou só o pode fazer de forma incompleta. Esta alteração genética, no caso mais simples, por exemplo, uma troca de um A por um C dentro do ADN, pode ser a causa causal de uma doença genética, se a função da célula num tecido for reduzida ou mesmo falhar como resultado, por exemplo
- Quando o metabolismo nas células funciona insuficientemente e os metabolitos se acumulam (por exemplo, defeito da lipoproteína lipase).
- Quando a síntese de blocos de construção básicos individuais pelo metabolismo é deficiente (por exemplo, defeito de glucose-6-fosfato desidrogenase).
- Se componentes individuais do sistema imunitário não funcionarem (por exemplo, Granulomatose Séptica ou Imunodeficiência Combinada Grave).
Num estudo recente, 4166 doenças raras e monogenéticas poderiam estar causalmente ligadas a 3163 genes [1]. Uma cura para a doença com uma causa genética só é possível se for possível compensar ou corrigir a causa causal.
Novas abordagens para a cura através da terapia genética
Se uma mutação genética claramente definida tiver sido identificada como a causa de uma doença, esta doença torna-se acessível para uma potencial terapia genética. Estão disponíveis vários procedimentos:
- Nas doenças em que a mutação genética leva à perda de expressão de proteínas ou à expressão de uma proteína defeituosa (“perda de função” ou mutações “disfuncionais”), a adição de genes pode ser realizada.
- Nas doenças em que a mutação genética leva a uma sobrefunção da proteína (mutações de “ganho de função”), pode ser tentada a reparação genética.
Além disso, o genoma da célula é estendido por um gene que codifica correctamente a proteína em falta ou em mau funcionamento e assim compensa a função do gene mutado. Isto resulta numa restauração da função original da proteína. A adição de genes tem sido testada em ensaios clínicos para o tratamento de numerosas doenças, com sucesso clínico para mais de 20 doenças hereditárias congénitas. Todos os produtos de terapia genética actualmente aprovados são baseados na adição de genes.
Na reparação de genes com nucleases como o CRISPR-Cas9 (fusão do Cas9 de nuclease com o CRISPR, Clustered Regularly Interspaced Short Palindromic Repeats), a sequência de ADN correcta é restaurada na célula, visando a mutação e corrigindo a mutação in situ (princípio: recortar e substituir). Assim, a função pretendida da proteína é restaurada e, subsequentemente, há uma normalização da função biológica. A utilização da reparação genética (“tesoura genética”/CRISPR-Cas) é possível independentemente da presença de mutações “ganho de função” ou “perda de função”. Tal modificação de informação genética específica da sequência é possível no laboratório, mas até agora só tem sido utilizada em oncologia no contexto de alguns poucos ensaios clínicos.
Genaddition
Os primeiros sucessos clínicos através da terapia genética foram alcançados no campo da imunologia. As causas das imunodeficiências congénitas residem nas células estaminais hematopoiéticas. Os doentes afectados podem, portanto, ser curados através do transplante de células estaminais hematopoiéticas de um dador não relacionado ou familiar (transplante alogénico). Os membros da família são raramente utilizados como doadores de HLA-haploidentical devido a uma taxa mais elevada de efeitos secundários. Os membros da família que têm uma identidade HLA mais elevada, mas que também são portadores do defeito genético, são mais susceptíveis de ser inelegíveis. Na ausência de um doador idêntico ao HLA (em cerca de 1/3 dos caucasianos, mais frequentemente noutros grupos étnicos), a terapia genética autóloga pode levar a uma cura da doença.
Para a terapia genética ex vivo, as células autólogas são esféricas após a mobilização com G-CSF, purificadas, enriquecidas e cultivadas ex vivo durante um curto período de tempo. Durante este período, as células estaminais são tratadas com o vector da terapia genética e depois reinfundidas no paciente, geralmente após quimioterapia para abater a medula óssea. Surpreendentemente, dependendo da imunodeficiência, a correcção parcial pode ser suficiente para uma melhoria clínica significativa.
A primeira publicação sobre terapia genética bem sucedida apareceu em 2000 sobre o tratamento de bebés com a forma mais grave de imunodeficiência congénita, imunodeficiência combinada grave, que geralmente leva à morte sem transplante no primeiro ano de vida devido às infecções mais graves [2].
Entre 2000 e 2006, todos os sucessos clínicos no campo da terapia genética foram alcançados com a ajuda dos chamados vectores retrovirais. Estes introduzem a sequência de correcção sob a forma de RNA nas células estaminais, onde é convertida em ADN por transcrição inversa e depois integrada no ADN do paciente (adição de genes). Posteriormente, formam-se proteínas funcionais e assim o defeito é clinicamente corrigido.
Para uma adição de genes in vivo, as células do corpo não são removidas, tratadas e devolvidas, mas as partículas virais com sequência de correcção são injectadas directamente no corpo. A primeira adição de genes in vivo foi publicada em 2007: para o tratamento da doença de Parkinson, os doentes foram injectados unilateralmente, subthalamicamente, com partículas virais adeno-associadas (AAV) modificadas que codificavam a descarboxilase do ácido glutâmico [3]. A grande maioria dos ensaios clínicos de terapia genética in vivo utiliza partículas modificadas de AAV para fornecer informação genética ao organismo. As excepções são o uso do vírus do herpes simples-1 modificado (HSV-1)- partículas virais derivadas do vírus [4] ou partículas adenovirais não replicantes [5] para o tratamento do glioblastoma e o teste de partículas virais derivadas do HIV-1 para o tratamento in vivo da doença de Parkinson [6].
Efeitos secundários nos primeiros estudos de terapia genética
Na primeira geração de terapias génicas ex vivocom vectores retrovirais γ, foi demonstrado que o sítio de integração do vector de terapia genética no genoma da célula alvo desempenha um papel fundamental no que diz respeito a possíveis efeitos secundários. Estes vectores de primeira geração integram preferencialmente sítios de início de transcrição próximos. Na primeira geração γ-retroviral de vectores da terapia genética, a expressão proteica foi impulsionada por um promotor no vector da terapia genética e melhorada por um melhorador. O melhorador do vector de terapia genética da primeira geração foi capaz de interagir com o sítio de início da transcrição do gene no qual o vector de terapia genética foi integrado. Se a integração num oncogene tivesse lugar, este oncogene poderia ser activado para além da expressão terapêutica da proteína, o que levou à cura da doença subjacente. Como resultado, as células estaminais em que esta constelação ocorreu tinham adquirido uma vantagem de crescimento e eram capazes de se multiplicar clonalmente. Em alguns ensaios clínicos com várias imunodeficiências, isto resultou em alguns doentes desenvolverem malignidades hematológicas como um efeito secundário da terapia genética [7–10]. Estes efeitos secundários levaram a uma mudança de vectores retrovirais γ para vectores lentivíricos derivados do vírus HIV-1 a partir de 2009, uma vez que o seu perfil de integração promete mais segurança [11]. Além disso, nos chamados vectores de auto-inactivação lentiviral (SIN), o promotor derivado do vírus HIV-1 e o seu elemento melhorador foram removidos. A ausência de elementos melhoradores nos vectores da segunda e terceira gerações de terapia genética impede a transacção de oncogenes mediada por melhoradores após tratamento ex vivo dascélulas estaminais. Até à data, os ensaios clínicos trataram as células estaminais de aproximadamente 100 pacientes com vectores de SIN lentiviral. Até agora, nenhum destes ensaios clínicos resultou no desenvolvimento de malignidades hematológicas, indicando uma melhoria significativa da segurança em comparação com o vector da primeira geração.
Os vectores de terapia genética baseados em AAV utilizados para a adição de genes in vivosó se integram em pequena medida no genoma das células alvo. Por conseguinte, a mutagénese de inserção é improvável. O maior potencial de risco dos vectores de terapia genética baseados em AAV reside em qualquer imunidade pré-existente às estruturas de superfície do vector AAV. Num ensaio clínico para o tratamento da hemofilia B, foi utilizado intramuscularmente um vector AAV2 codificador de factor de coagulação do sangue IX. Uma resposta imunitária mediada por células T levou à eliminação de todas as células geneticamente modificadas e pôs fim ao efeito terapêutico [12]. Em contraste, a administração intravenosa resultou na absorção dos vectores em hepatócitos e efeitos terapêuticos a longo prazo em doentes nos quais não foi possível detectar anticorpos contra AAV antes da inclusão no estudo [13].
Sucessos da terapia genética
A primeira geração de vectores de terapia genética γ-retroviral forneceu provas de que a terapia genética pode levar ao sucesso clínico da X-SCID (imunodeficiência combinada grave ligada ao X) [2], ADA (adenosina deaminase)-SCID [14], X-CGD (granulomatose séptica ligada ao X) [9], epidermólise bolhosa [15] e para a síndrome de Wiskott Aldrich (WAS) [16].
Devido aos efeitos secundários acima mencionados no tratamento de X-SCID, X-CGD e WAS com vectores de terapia genética de primeira geração (γ-vectores retrovirais com promotor/reforçador completo), foram desenvolvidos vectores de SIN lentiviral, levando a uma história clínica de sucesso. Até agora, os vectores do SIN lentiviral alcançaram sucesso clínico na terapia genética ex vivo para o tratamento de
- Adrenoleucodistrofia ligada ao X (ALD) [17]
- γ-Thalassemia [18,19]
- O QUE [20]
- X-SCID [21]
- ADA-SCID [22]
- Doença das células falciformes [23]
- COL7A1 Epidermólise Bullosa [24]
- Leucodistrofia metacromática [25]
Entretanto, a terapia genética in vivomediada por AAV também tem sido utilizada no tratamento de pelo menos 14 indicações:
Neurologia
- Parkinson [3,26,27]
Oftalmologia
- Congregação do fígado. Amaurose [28,29]
- Chorioideremia [30,31]
- Degeneração macular relacionada com a idade [32]
- A neuropatia óptica hereditária de Leber [33,34]
Hematologia
- Hemofilia B [13]
Distrofia muscular
- Distrofia muscular Becker [35]
- Atrofia muscular espinhal tipo I (SMA1) [36]
Doenças metabólicas
- Doença de Pompe [37]
- α-1-antitripsina (AAT) deficiente [38,39]
- Mucopolissacaridose tipo IIIB [40]
- Deficiência de descarboxilase de aminoácidos aromáticos [41,43]
- Deficiência de lipoproteína lipase [42]
No tratamento da leucemia, as células CAR-T são utilizadas em numerosos estudos e agora também em terapias aprovadas. Para este fim, os receptores quiméricos de antigénios são introduzidos em células T autólogas de doentes afectados. Isto permite que estas células T geneticamente modificadas (células CAR-T) reconheçam e eliminem células tumorais [44–46].
Reparação de género
Até agora, não tem sido possível ter vectores de terapia genética lenti- ou γ-retroviral integrados em sítios pré-determinados do genoma. A inserção e/ou correcção específica da sequência só se tornou possível depois de ter sido possível cortar o ADN de uma forma específica da sequência. Isto é feito usando nucleases como o CRISPR-Cas. Neste sistema de terapia genética, foi produzida uma fusão das Repetições Palindrómicas Curtas Interespaçadas Regularmente (CRISPR) e do Cas9 de corte de ADN (CRISPR/Cas9) [47]. A quebra resultante no ADN pode então ser reparada pela célula de duas maneiras diferentes: Ou as extremidades são reconectadas num processo defeituoso chamado de junção final não-homológica. O segundo mecanismo de reparação é a recombinação homóloga. Este caminho é tomado quando há ADN reparado na célula cujas extremidades coincidem com a sequência de ADN no local da quebra de ADN. Este ADN reparador é administrado às células juntamente com o nuclease e serve como modelo genético. Na recombinação homóloga, a quebra do ADN é então fechada pelas enzimas de reparação celular endógena de acordo com a sequência de ADN do ADN de reparação. Isto torna possível introduzir um gene de correcção no genoma de uma forma específica de sequência ou corrigir geneticamente mutações pontuais. A adição de genes específicos da sequência ainda não encontrou aplicação clínica porque a eficiência do método tem sido limitada até agora. Contudo, os recentes desenvolvimentos técnicos estão a trazer os ensaios clínicos para o reino da possibilidade.
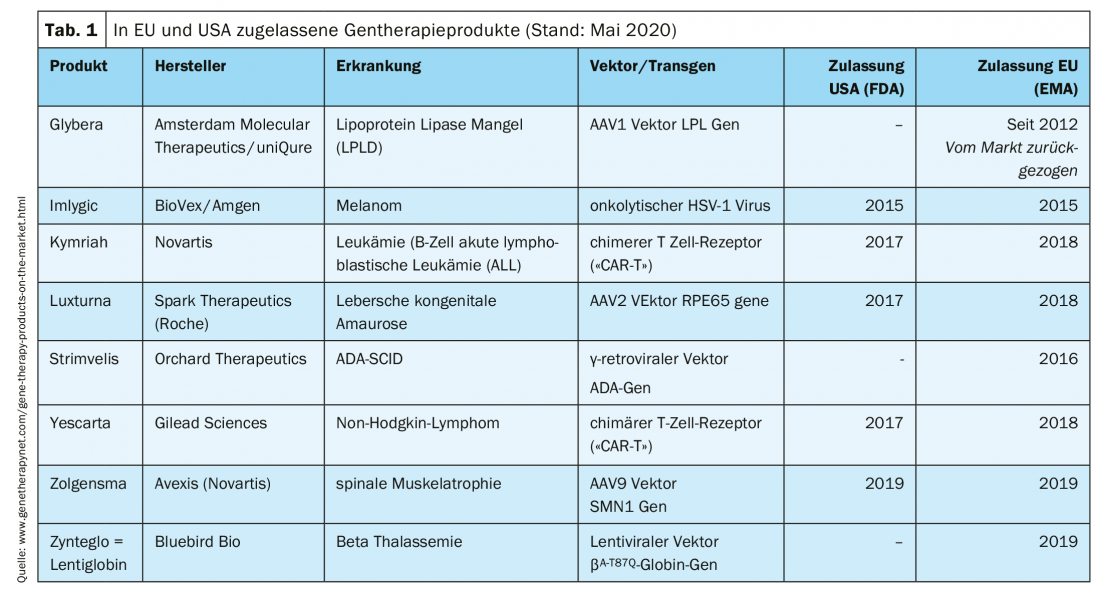
Produtos de terapia genética clinicamente aprovados
O tratamento de pacientes com produtos de terapia genética é possível em centros especializados. Vários produtos já receberam autorização de comercialização (Tab. 1).
Mensagens Take-Home
- A terapia genética como tratamento causal para doenças hereditárias monogenéticas está cada vez mais a encontrar o seu caminho para a clínica.
- Utilizando vectores virais, a terapia genética baseada na adição de genes tem sido utilizada com sucesso em ensaios clínicos para certas doenças hematológicas desde 2000.
- doenças e imunodeficiências congénitas.
- Até à data, a reparação de genes alvo usando nucleases tem sido utilizada em poucos estudos para doenças oncológicas em que a função de um gene deve ser desligada.
- Algumas doenças podem ser tratadas utilizando células T geneticamente modificadas (células CAR-T).
- A terapêutica genética já está actualmente disponível como produtos aprovados para 7 doenças.
Literatura:
- Ehrhart F, et al: History of rare diseases and their genetic causes – a data driven approach. bioRxiv 2020; preprin doi: https://doi.org/10.1101/595819
- Cavazzana-Calvo, M et al: Gene Therapy of Human Severe Combined Immunodeficiency (SCID)-X1 Disease. Science 2000; 288: 669-672.
- Kaplitt MG, et al: Safety and Tolerability of Gene Therapy With an Adeno-Associated Virus (AAV) Borne GAD Gene for Parkinson’s Disease: An Open Label, Phase I Trial. Lancet 2007; 369(9579): 2097-2105.
- Todo T: Imunoterapia Activa: Vírus Oncolítico Terapia Usando HSV-1 Adventário Exp Med Biol. 2012; 746: 178-186.
- Brenner AJ, et al: Safety and Efficacy of VB-111, anticancer Gene Therapy, in Patients With Recurrent Glioblastoma: Results of a Phase I/II Study. Neuro Oncol 2020; 22(5): 694-704.
- Palfi S, et al: Acompanhamento a Longo Prazo de um Estudo de Fase I/II de ProSavin, uma Terapia de Vector Gene Lentiviral para a Doença de Parkinson. Hum Gene Ther Clin Dev 2018; 29(3): 148-155.
- Hacein-Bey-Abina S, et al: Serious Adverse Event After Successful Gene Therapy for X-linked Severe Combined Immunodeficiency. N Engl J Med 2003; 348(3): 255-256.
- Aiuti A, et al.: The Committee for Advanced Therapies’ of the European Medicines Agency Reflection Paper on Management of Clinical Risks Deriving from Insertional Mutagenesis. Human Gene Ther Clin Dev 2013; 24: 47-54.
- Ott MG, et al: Correcção da doença granulomatosa crónica ligada ao X pela Terapia Genética, Aumentada pela Activação Insercional de MDS1-EVI1, PRDM16 ou SETBP1. Nat Med 2006; 12(4): 401-409.
- Siler U, et al.: Combinação bem sucedida de Terapia Sequencial Genética e Resgate Allo-HSCT em Duas Crianças com X-CGD – Importância do Tempo. Moeda Gene Ther 2015; 15(4): 416-427.
- Serrao E, et al: Sítios de Integração de DNA Retroviral: Da Investigação Básica a Aplicações Clínicas. AN Critérios Rev Biochem Mol Biol 2015; 28: 1-17.
- Manno CS, et al: Transdução bem sucedida do fígado em hemofilia por AAV-Factor IX e Limitações Impostas pela Resposta Imune do Hospedeiro. Nat Med 2006; 12: 342-347.
- Nathwani AC, et al: Long-term Safety and Efficacy of Factor IX Gene Therapy in Hemophilia B. N Engl J Med 2014; 371(21): 1994-2004.
- Aiuti A, et al.: Correcção da ADA-SCID por Terapia com Gene de Células-Tronco Combinada com Condicionamento Não-Mieloablativo. Science 2002; 296(5577): 2410-2413.
- Mavilio F, et al: Correcção da Epidermólise Bullosa Juncional por Transplante de Células-Tronco Epidérmicas Geneticamente Modificadas. Nat Med 2006; 12(12): 1397-1402.
- Boztug K, et al: Stem-cell Gene Therapy for the Wiskott-Aldrich Syndrome. N Engl J Med 2010; 363(20): 1918-1927.
- Cartier N, et al.: Hematopoietic Stem Cell Gene Therapy With a Lentiviral Vector in X-linked Adrenoleukodystrophy. Ciência 2009; 326(5954): 818-823.
- Cavazzana-Calvo M, et al.: Transfusion Independence and HMGA2 Activation After Gene Therapy of Human β-Thalassaemia. Natureza 2010; 467(7313): 318-322.
- Thompson AA, et al: Gene Therapy in Patients with Transfusion-Dependent β-Thalassemia. N Engl J Med 2018; 378: 147993.
- Aiuti A, et al.: Terapia génica hematopoiética lentiviral em doentes com síndrome de Wiskott-Aldrich. Ciência 2013; 341(6148): 1233151.
- De Ravin SS, et al: Lentiviral Hematopoietic Stematopoietic Gene Therapy for X-linked Severe Combined Immunodeficiency. Sci Transl Med 2016; 8(335): 335ra57.
- Mullard A: EMA greenlights segunda terapia genética. Nat Rev Drug Discov 2016; 15: 299.
- Ribeil JA, et al: Terapia Genética num Paciente com Doença Falciforme. N Engl J Med 2017; 376(9): 848-855.
- Lwin SM, et al: Safety and Early Efficacy Outcomes for Lentiviral Fibroblast Gene Therapy in Recessive Dystrophic Epidermolysis Bullosa. JCI Insight 2019; 4(11): e126243.
- Biffi A, et al.: A terapia genética das células estaminais hematopoiéticas lentiviral beneficia a leucodistrofia metacromática. Ciência 2013; 341(6148): 1233158.
- Niethammer M, et al: Gene Therapy Reduz os Sintomas da Doença de Parkinson através da Reorganização da Conectividade Cerebral Funcional. Sci Transl Med 2018; 10(469): eaau0713.
- Heiss JD, et al: Ensaio de Ressonância Magnética Orientada por Putaminal Gene Terapia para a Doença de Parkinson Avançada. Mov Disord 2019; 34(7): 1073-1078.
- Bainbridge JWB, et al: Effect of Gene Therapy on Visual Function in Leber’s Congenital Amaurosis. N Engl J Med 2008; 358(21): 2231-2239.
- Bainbridge JWB, et al: Long-term Effect of Gene Therapy on Leber’s Congenital Amaurosis. N Engl J Med 2015; 372(20): 1887-1897.
- MacLaren RE, et al: Retinal Gene Therapy in Patients With Choroideremia: Initial Findings From a Phase 1/2 Clinical Trial. Lanceta 2014; 383: 1129-1137.
- Xue K, et al: Efeitos benéficos na visão em pacientes sob terapia de retina para a coroideremia. Nat Med 2018; 24(10): 1507-1512.
- Rakoczy EP, et al: Gene Therapy With Recombinant Adeno-Associated Vectors for Neovascular Age-Related Macular Degeneration: 1 Year Follow-Up of a Phase 1 Randomised Clinical Trial. Lancet 2015; 386(10011): 2395-2403.
- Feuer WJ, et al: Gene Therapy for Leber Hereditary Optic Neuropathy: Initial Results. Oftalmologia 2015; 123(3): 558-570.
- Bouquet C, et al: Immune Response and Intraocular Inflammation in Patients With Leber Hereditary Optic Neuropathy Treated With Intravitreal Injection of Recombinant Adeno-Associated Virus 2 Carrying the ND4 Gene: A Secondary Analysis of a Phase 1/2 Clinical Trial. JAMA Ophthalmol 2019; 137(4): 399-406.
- Mendell JR, et al: A Phase 1/2a Follistatin Gene Therapy Trial for Becker Muscular Dystrophy. Mol Ther 2015; 23(1): 192-201.
- Mendell JR, et al: Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. N Engl J Med 2017; 377(18): 1713-1722.
- Corti M, et al: B-Cell Depletion Is Protective Against Anti-AAV Capsid Immune Response: A Human Subject Case Study. Mol Ther Methods Clin Dev 2014; 1: 14033.
- Calcedo R, et al: Class I-restricted T-cell Responses to a Polymorphic Peptide in a Gene Therapy Clinical Trial for α-1-antitrypsin Deficiency. Proc Natl Acad Scien U S A 2017; 114(7): 1655-1659.
- Mueller C, et al.: Ano de Expressão e Reparação de Defeitos Neutrófilos após Terapia Genética na Deficiência de Antitripsina Alfa-1. Mol Ther 2017; 25(6): 1387-1394.
- Tardieu M, et al: Intracerebral Gene Therapy in Children With Mucopolysaccharidosis Type IIIB Syndrome: An Uncontrolled Phase 1/2 Clinical Trial. Lancet Neurol 2017; 16(9): 712-720.
- Chien YH, et al: Eficácia e Segurança da Terapia Genética AAV2 em Crianças com Deficiência de Ácido L-aminoácido Aromático Descarboxilase: Um rótulo aberto, Fase 1/2 de ensaio. Lancet Child Adolesc Health 2017; 1(4): 265-273.
- Kassner U, et al.: Gene Therapy in Lipoprotein Lipase Deficiency: Relatório de caso sobre o primeiro paciente tratado com Alipogene Tiparvovec em condições de prática diária. Hum Gene Ther 2018; 29(4): 520-527.
- Kojima K, et al: Gene Therapy Improves Motor and Mental Function of Aromatic L-Amino Acid Decarboxylase Deficiency. Brain 2019; 142(2): 322-333.
- Porter DL, et al: Receptor de Antigénio Quimérico – Células T Modificadas em Leucemia Linfóide Crónica. N Engl J Med 2011; 365(8): 725-733.
- Savoldo B, et al: CD28 Costimulation Improves Expansion and Persistence of Chimeric Antigen Receptor-Modified T Cells in Lymphoma Patients. J Clin Invest 2011; 121(5): 1822-1826.
- Grupp SA, et al: Chimeric Antigen Receptor-Modificado de Células T para Leucemia Linfóide Aguda. N Engl J Med 2013; 368(16): 1509-1518.
- Yang G, Huang X: Métodos e Aplicações do CRISPR/Cas System for Genome Editing in Stem Cells. Cell Regen (Lond) 2019; 8(2): 33-41.
PRÁTICA DO GP 2020; 15(9): 6-10









