O tratamento cirúrgico de doentes com doenças do tecido conjuntivo hereditário tem como objectivo prevenir a dissecção da aorta. Porque isto é responsável pela elevada taxa de mortalidade da população de doentes. Quando é dada a indicação? E que opções de terapia medicamentosa existem?
As doenças do tecido conjuntivo hereditário com manifestações vasculares dificilmente estão presentes na consciência do clínico geral. Durante muito tempo, a síndrome de Marfan foi o único diagnóstico diferencial nesta direcção em doentes jovens com um evento da aorta torácica. Centenas de genes associados a formas sindrómicas e não sindrómicas de doenças da aorta foram identificados na última década [1].
Além disso, porém, quase metade dos doentes só são diagnosticados no contexto de uma complicação vascular. Enquanto os aneurismas são geralmente detectados durante um exame de rotina, a dissecção da aorta é uma das emergências cirúrgicas associadas a uma morbilidade e mortalidade elevadas.
A síndrome de Marfan é uma desordem autossómica dominante com uma incidência de aproximadamente 1:5000 nascidos vivos. As manifestações oculares, esqueléticas e cardíacas típicas são causadas por mutações no gene da fibrilina-1, que levam à sobreativação do caminho de sinalização TGFβ [2].
Bart Loeys e Hal Dietz identificaram há dez anos uma subpopulação de doentes que se destacavam pela sua úvula dividida, grande distância interocular e vasos tortuosos. Entretanto, foram identificados vários genes que levam a um fenótipo do grupo da síndrome de Loeys-Dietz. A identificação destes pacientes é importante porque as dissecções ocorrem frequentemente em diâmetros da aorta que não eram anteriormente considerados indicações para a substituição profiláctica da aorta [3].
A rara síndrome vascular de Ehlers-Danlos é causada por mutações no gene que codifica o colagénio III. Estes pacientes são notáveis pela sua elevada taxa de dissecação e ruptura sem formação prévia de aneurisma. Isto torna os cuidados destes pacientes muito difíceis. A sobrevivência média é de 48 anos se não for tratada, com os primeiros acontecimentos a ocorrerem normalmente na terceira a quarta década de vida [4].
Opções de tratamento cirúrgico
O objectivo do tratamento cirúrgico de pacientes com doenças do tecido conjuntivo hereditário é evitar a dissecção da aorta devido à dilatação da aorta, uma vez que esta é responsável pela elevada mortalidade nesta população de pacientes. As directrizes de 2014 da Sociedade Europeia de Cardiologia (ESC) fazem uma distinção clara na indicação de substituição aórtica electiva entre pacientes Marfan ou Loeys-Dietz e pacientes sem doença genética subjacente [5]. A dificuldade aqui reside na demarcação. Em pacientes jovens, a presença de um componente genético deve ser assumida. Em pacientes sem doença do tecido conjuntivo, recomenda-se a substituição profilática da aorta quando o diâmetro da raiz da aorta ou aorta ascendente for 55 mm. Os doentes com Marfan, por outro lado, devem ser operados a 50 mm, e no caso de factores de risco existentes (tais como dissecções na família) mesmo a partir de 45 mm. Em doentes de Loeys-Dietz, a substituição electiva é aconselhada a 42-45 mm. Em regra, a raiz aórtica é substituída principalmente. Se possível, é feita uma tentativa de preservar a própria válvula do paciente. Se tal não for viável devido a danos na válvula, é implantada uma conduta de suporte de válvula.
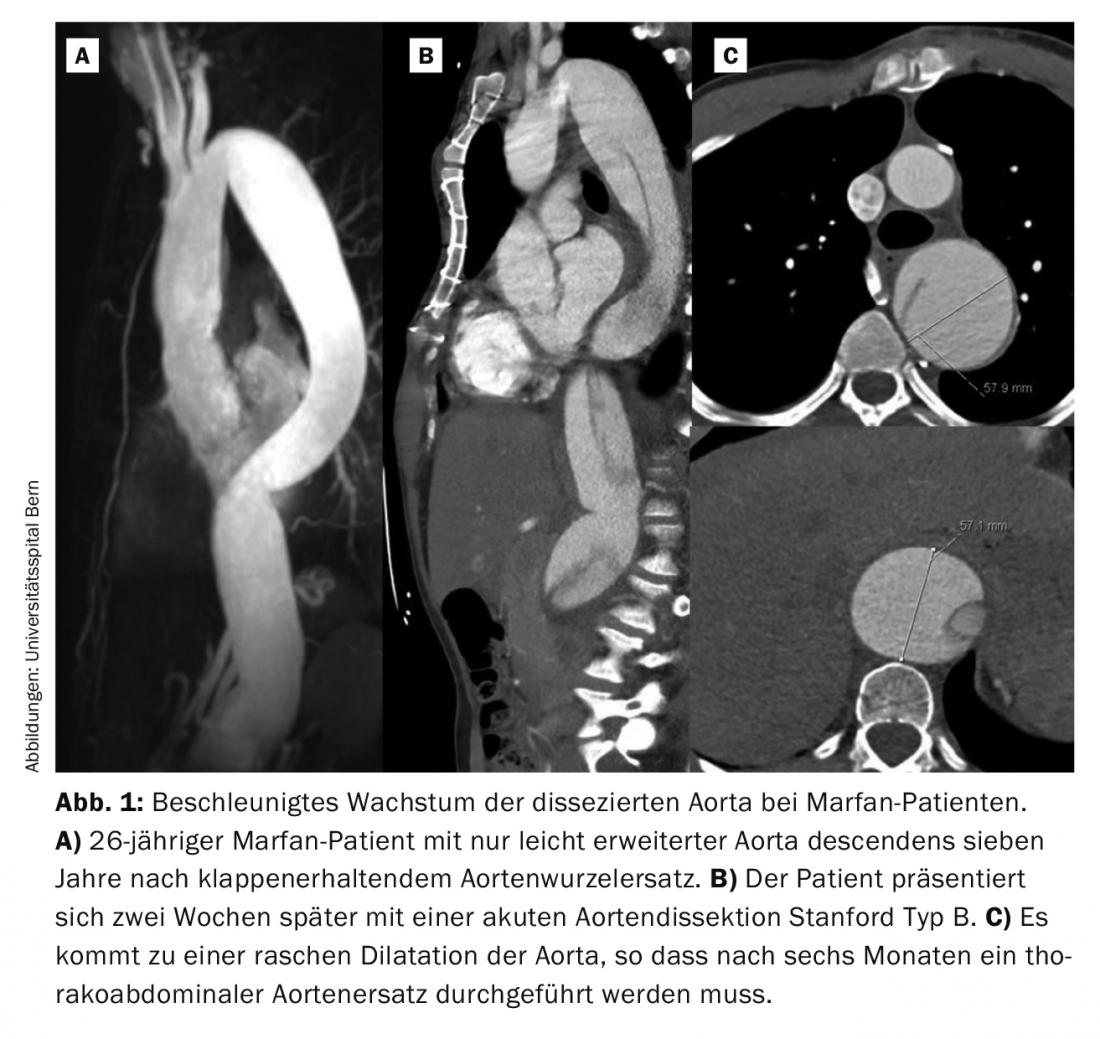
Contudo, muitas vezes a manifestação inicial é a dissecção aguda da aorta de Stanford tipo A ou B. Na nossa experiência ao longo dos últimos vinte anos, um terço dos pacientes apresentou dissecção aguda. A dissecção aguda tipo A é uma emergência cirúrgica e deve ser tratada imediatamente. Mesmo com uma cirurgia bem sucedida, metade destes pacientes requerem uma reoperação, na sua maioria na aorta distal, ou seja, não substituída. Em pacientes com substituição eletiva da raiz da aorta, apenas cerca de 10% dos pacientes necessitam de uma repetição da operação. Na maioria dos casos, trata-se de pacientes que sofreram entretanto uma dissecção da aorta de tipo B. Embora a dissecção da aorta tipo B seja frequentemente inicialmente “sem complicações”, ou seja, sem má perfusão, a aorta deve ser posteriormente substituída toracoabdominalmente na grande maioria dos doentes com Marfan. Uma característica típica dos doentes com Marfan é o rápido crescimento da aorta dissecada nas primeiras semanas e alguns meses (Fig. 1) . A experiência mostra que 50% dos pacientes necessitam de cirurgia no primeiro ano após o evento [6].
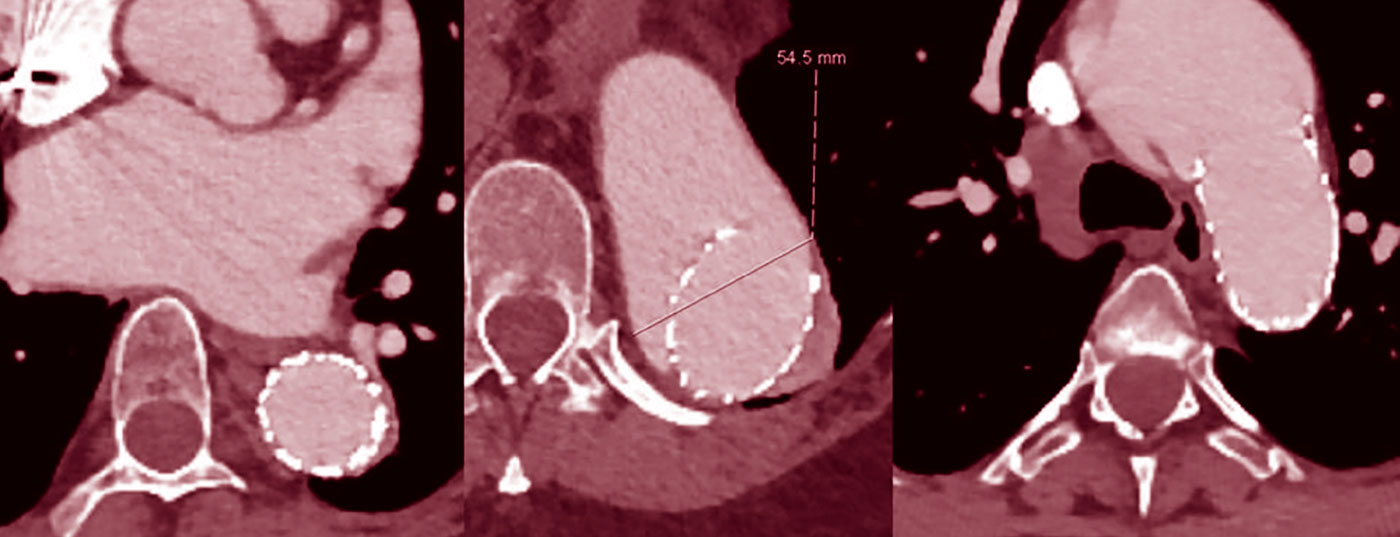
O tratamento dos segmentos dilatados da aorta toracoabdominal com endopróteses não é geralmente recomendado porque os segmentos das embarcações da zona de aterragem se dilatam frequentemente em segundo lugar (Fig. 2), mesmo que inicialmente haja uma boa remodelação [7].

Opções de medicamentos
A dilatação e o risco resultante de dissecção da aorta é a principal causa de morbilidade e mortalidade em doentes com doenças hereditárias do tecido conjuntivo. O objectivo do tratamento medicamentoso é reduzir a progressão da dilatação e a ocorrência de dissecções. A redução da pressão arterial para valores sistólicos máximos de 120 mmHg e a taxa de aumento da pressão na região aórtica com cada batimento cardíaco são de importância relevante. Tradicionalmente, o beta-bloqueador é a droga de eleição [8]. Estudos demonstraram uma taxa de dilatação mais baixa em doentes em terapia com beta-bloqueadores, mas sem significado clínico em termos de sobrevivência [9].
Nos últimos anos, o bloqueador de receptores losartan ATII tem sido estudado com mais detalhe. Como interfere directamente com a via de sinalização TGFβ , depositam-se grandes esperanças no seu efeito no que respeita à prevenção da dilatação e dissecção da aorta [10]. No entanto, um grande ensaio aleatório em crianças com síndrome de Marfan não encontrou diferença entre os beta-bloqueadores e o losartan [11]. O estudo foi controverso porque acabou por comparar uma dose muito elevada de atenolol com uma dose relativamente baixa de losartan. Devido ao perfil de efeito secundário inferior, o antagonista ATII é, portanto, a nossa primeira escolha para o tratamento de pacientes ingénuos com função de bomba normal. Os antagonistas da ATII são bem tolerados, especialmente por crianças e adolescentes. Estudos mais pequenos mostraram que a tensão arterial não é significativamente reduzida nesta população de doentes [12].
Controlo do progresso
A fim de detectar atempadamente a ocorrência ou progressão de um aneurisma da aorta e evitar a dissecção da aorta por substituição profiláctica precoce da aorta, é essencial efectuar um acompanhamento atento [13]. É importante avaliar sistematicamente todos os segmentos da aorta. Além disso, os controlos após cirurgia da aorta são importantes para detectar possíveis complicações como a formação de aneurisma noutras secções da aorta e dissecções assintomáticas numa fase precoce.
Nas nossas horas de consulta, os pacientes são verificados após três e doze meses por meio de angio-TC e mais tarde, dependendo dos resultados, uma ou três vezes por ano. Para reduzir a dose cumulativa de radiação, isto deve ser feito principalmente por ressonância magnética. O exame MRI também oferece a vantagem da imagem funcional no que diz respeito à função da bomba, viciação das válvulas e diagnóstico de isquemia. As operações anteriores com implantes raramente constituem um problema ao avaliar as imagens. A angiografia CT ainda é actualmente superior à ressonância magnética na avaliação das dissecções. Os controlos ecocardiográficos são efectuados no caso de vitia ou st.n. Substituição de válvulas realizada uma vez por ano ou alternadamente com exame de ressonância magnética.
Uma situação especial nos cuidados a longo prazo de pacientes com doenças hereditárias do tecido conjuntivo é o desejo de ter filhos. Há aqui poucas provas e as recomendações das sociedades profissionais são parcialmente contraditórias [5,14,15]. Em geral, um diâmetro aórtico de <40 mm é considerado um risco aceitável de engravidar sob controlo ecocardiográfico próximo e bloqueio beta. Para diâmetros >45 mm, a cirurgia profiláctica é claramente aconselhada. Para diâmetros entre 40 e 45 mm, a situação individual do paciente deve ser pesada com muito cuidado. O factor de risco aqui é certamente um historial familiar positivo no que diz respeito às dissecções, especialmente durante a gravidez.
As directrizes actuais recomendam o rastreio de todos os parentes de primeiro grau num doente com aneurisma da aorta torácica. Este passo pode contribuir significativamente para reduzir a taxa de pacientes com dissecções.
Mensagens Take-Home
- Em doentes com doenças hereditárias do tecido conjuntivo, a indicação para a substituição profilática da aorta já é dada com diâmetros de 45-50 mm.
- A avaliação regular e sistemática de todos os segmentos da aorta previne dissecções e rupturas.
- Todos os familiares de 1º grau de pacientes com aneurisma da aorta torácica devem ser rastreados para detectar a presença de um.
Literatura:
- Brownstein AJ, et al: Genes associados ao aneurisma e dissecção da aorta torácica: actualização de 2018 e implicações clínicas. Aorta (Stamford) 2018; 6: 13-20.
- Habashi JP, et al: Losartan, um antagonista de AT1, previne o aneurisma da aorta num modelo de rato da síndrome de Marfan. Science 2006; 312: 117-121.
- Loeys BL, et al: Sindromes de aneurisma causados por mutações no receptor TGF-beta. N Engl J Med 2006; 355: 788-798.
- Pepin M, et al: Características clínicas e genéticas da síndrome de Ehlers-Danlos tipo IV, o tipo vascular. N Engl J Med 2000; 342: 673-680.
- Erbel R, et al.: 2014 ESC Guidelines on the diagnosis and treatment of aortic diseases: Document covering acute and chronic aortic diseases of the thoracic and abdominal aorta of the adult. A Task Force para o Diagnóstico e Tratamento das Doenças da Aorta da Sociedade Europeia de Cardiologia (ESC). Eur Heart J 2014; 35: 2873-2926.
- Schoenhoff FS, et al: A dissecção aguda da aorta determina o destino dos segmentos aórticos inicialmente não tratados na síndrome de Marfan. Circulação 2013; 127: 1569-1575.
- Grabenwöger M, et al: Thoracic Endovascular Aortic Repair (TEVAR) for the treatment of aortic diseases: a position statement from the European Association for Cardio- Thoracic Surgery (EACTS) and the European Society of Cardiology (ESC), in collaboration with the European Association of Percutaneous Cardiovascular Interventions (EAPCI). Eur Heart J 2012; 33: 1558-1563.
- Shores J, et al: Progressão da dilatação da aorta e o benefício do bloqueio beta-adrenérgico a longo prazo na síndrome de Marfan. N Engl J Med 1994; 330: 1335-1341.
- Gersony DR, et al: O efeito da terapia com beta-bloqueador no resultado clínico em pacientes com síndrome de Marfan: uma meta-análise. Int J Cardiol 2007; 114: 303-308.
- Habashi JP, et al.: Angiotensina II tipo 2 receptor de sinalização atenua o aneurisma da aorta em ratos através do antagonismo ERK. Ciência 2011; 332: 361-365.
- Lacro RV, et al: Atenolol versus losartan em crianças e jovens adultos com síndrome de Marfan. N Engl J Med 2014; 371: 2061-2071.
- Brooke BS, et al: bloqueio de Angiotensina II e dilatação da raiz aórtica na síndrome de Marfan. N Engl J Med 2008; 358: 2787-2795.
- Jondeau G, et al: Aortic event rate in the marfan population a cohort study. Circulação 2012; 125: 226-232.
- Baumgartner H, et al: Orientações CES para a gestão das doenças cardíacas congénitas em adultos (nova versão 2010). Eur Heart J 2010; 31: 2915-2957.
- Hiratzka LF, et al.2010 ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM guidelines for the diagnosis and management of patients with Thoracic Aortic Disease: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines, American Association for Thoracic Surgery, American College of Radiology, American Stroke Association, Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, Society of Interventional Radiology, Society of Thoracic Surgeons, and Society for Vascular Medicine. Circulação 2010; 121: e266-369.
CARDIOVASC 2018; 17(5): 27-29









