Os termos doença pulmonar intersticial ou doença parenquimatosa difusa referem-se a um grupo de patologias que afectam o epitélio dos alvéolos (epitélio alveolar), o endotélio dos capilares pulmonares, a membrana basal ou os tecidos perivasculares e perilinfáticos do pulmão. A fibrose pode desenvolver-se no decurso da maioria das ILDs.
Os termos doença pulmonar intersticial ( [ILD]) ou doença pulmonar parenquimatosa difusa ( [DPLD]) referem-se a um grupo de patologias que afectam o epitélio dos alvéolos, o endotélio dos capilares pulmonares, a membrana basal ou os tecidos perivasculares e perilinfáticos do pulmão. Bronquíolos e brônquios são também frequentemente afectados. As DPIs não incluem, por exemplo, doenças obstrutivas das vias respiratórias ou pneumonia directamente relacionada com agentes patogénicos. A fibrose pode desenvolver-se no decurso da maioria das ILDs.
Divisão
O grupo de ILDs é heterogéneo e pode ser subdividido de diferentes formas. A divisão mais simples é nas chamadas pneumonias intersticiais idiopáticas (IIPs), por um lado, e nas ILDs com uma causa conhecida, por outro. A causa da DPI pode ser, por exemplo, doenças sistémicas como a sarcoidose ou colagenoses, bem como influências externas como a alveolite alérgica exógena (AEA), as pneumoconiose de toxicidade medicamentosa ou a pneumonite radiogénica.
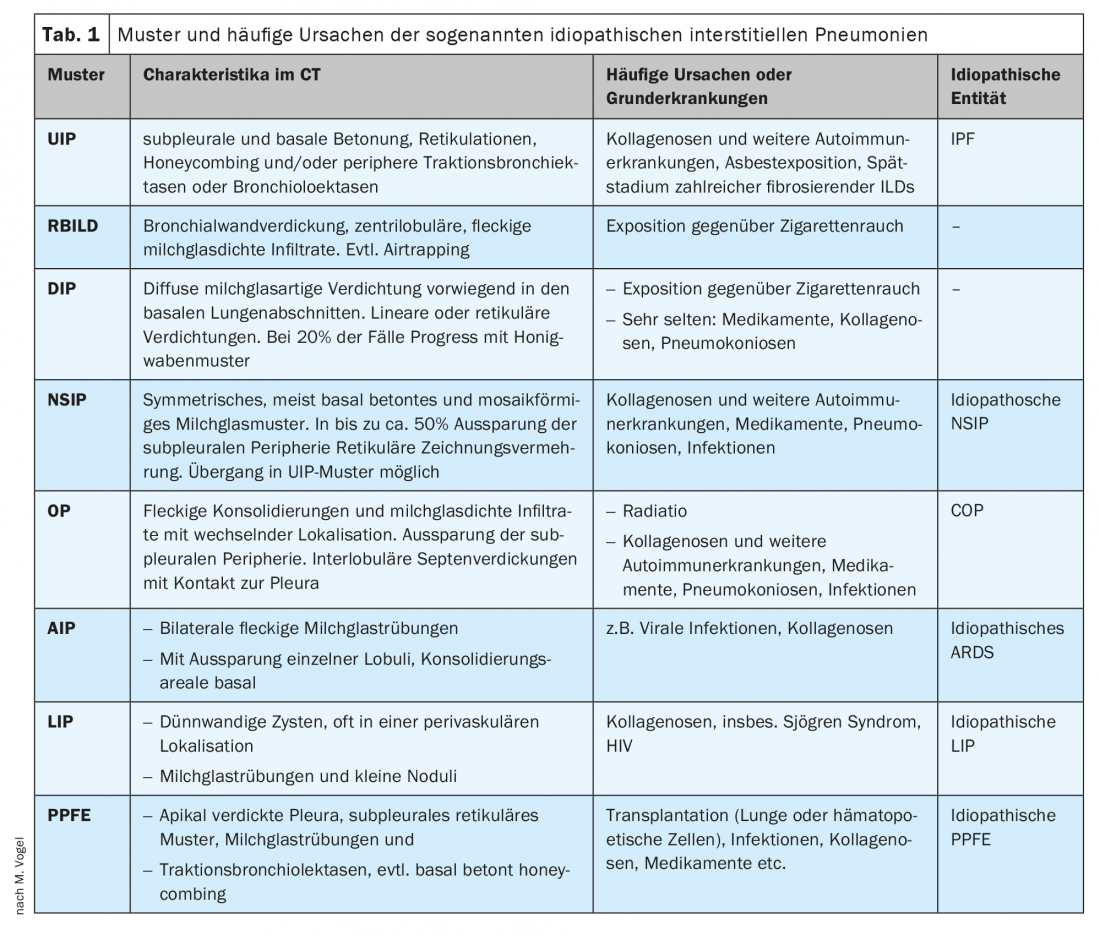
Entre os PIIs, oito padrões e entidades associadas distinguem-se predominantemente no CT de fatias finas e no espécime histológico:
- O padrão da habitual pneumonia intersticial (PIU) na fibrose pulmonar idiopática (IPF).
- O padrão da bronquiolite respiratória (RB) na bronquiolite respiratória associada à doença intersticial pulmonar (RBILD).
- O padrão de pneumonia intersticial despamativa (DIP) em DIP com o mesmo nome.
- O padrão NSIP em pneumonia intersticial não específica (NSIP).
- O padrão de organização da pneumonia (OP) na pneumonia criptogénica organizadora (COP).
- O padrão da pneumonia intersticial aguda (AIP) com o quadro clínico da SDRA.
- O padrão LIP na pneumonia intersticial linfóide (LIP).
- O padrão da fibroelastose pleuroparenquimatosa na doença homónima (PPFE).
Estes padrões não são sinónimos de uma doença. Pelo contrário, por um lado, podem ser idiopáticas ou, por outro lado, podem estar presentes várias causas conhecidas: Fumo de cigarro para RBILD e DIP e, entre outros, colagenoses para UIP, NSIP, OP, LIP e AIP [1] ou pneumonias virais para AIP, NSIP e OP (ver Tabela 1 para padrões e causas comuns). Não existe uma relação clara entre padrão e doença desencadeante: a mesma doença pode desencadear padrões diferentes em pacientes diferentes. Além disso, os padrões mostram um espectro diferente com variações parcialmente grandes e pontos em comum com outros padrões. O padrão de ouro para o diagnóstico de DPIs é, portanto, o consenso interdisciplinar de pelo menos pneumologia, radiologia e patologia [2,3]. Outras disciplinas especializadas, como a reumatologia, devem também ser consultadas regularmente. Embora o padrão na tomografia computorizada de camada fina seja geralmente indicativo, este artigo mostrará a importância de todas as disciplinas relevantes e especialmente a informação anamnéstica e clínica de todo o curso da doença para o diagnóstico. O foco aqui será a classificação do padrão UIP.
Padrão UIP e IPF
O IPF é o mais comum e, com uma média de sobrevivência de 3-4 anos, o PII mais desfavorável em termos de prognóstico. A prevalência é de 2-29 por 100 000, a incidência de cerca de 10 por 100 0000/ano [4]. A terapia com inibidores específicos de fibroblastos pode atrasar a progressão mas não reverter a doença [4]. A terapia anti-inflamatória, que é indicada para muitas outras PII, tem um impacto negativo no prognóstico [5]. O padrão UIP presente no IPF também pode ser não-idiopático, por exemplo, no contexto de colagenoses, fibrosing EAA [6], após exposição ao amianto ou sarcoidose.
Uma constelação típica para o IPF é a seguinte: Sexo masculino, idade superior a 60 anos e história de tabagismo, que também pode remontar a vários anos ou, alternativamente, a um aumento da ocorrência familiar do IPF [3,7]. Se nenhuma doença ou influência subjacente correspondente puder ser identificada num paciente com um padrão UIP, é feito o diagnóstico de IPF. Esta é uma doença independente e tratável cuja génese parece ser multifactorial. O “padrão UIP típico” (categoria 1) é o CT morfologicamente caracterizado pela ênfase subpleural e basal das alterações fibróticas. Encontram-se reticulações e uma estrutura pulmonar tipo favo de mel, o chamado padrão alveolar, com ou sem bronquiectasia de tracção periférica ou bronquioloectasia. A assimetria ocorre em cerca de 25% [8].
Nem todos os casos de IPF mostram as típicas alterações morfológicas: Se apenas falta o padrão alveolar, fala-se de um “provável padrão IPF” (categoria 2). Além disso, a informação clínica não está normalmente disponível na sua totalidade quando a tomografia computorizada é realizada. Em primeiro lugar, portanto, a certeza ou probabilidade de um padrão UIP é determinada com base nas imagens de TC finamente reconstruídas. Foram definidos novos critérios para este fim em 2018 (Tab. 2) [9].
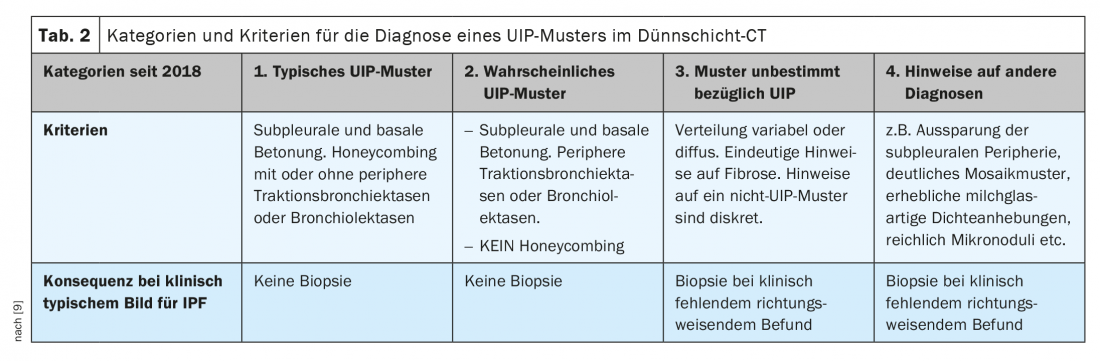
A consequência da classificação numa das categorias é a orientação para o procedimento de diagnóstico posterior. Num contexto clínico típico do IPF, a biopsia pulmonar não é realizada para as categorias 1 e 2. [3,7,9].
Categoria 1: padrão UIP típico
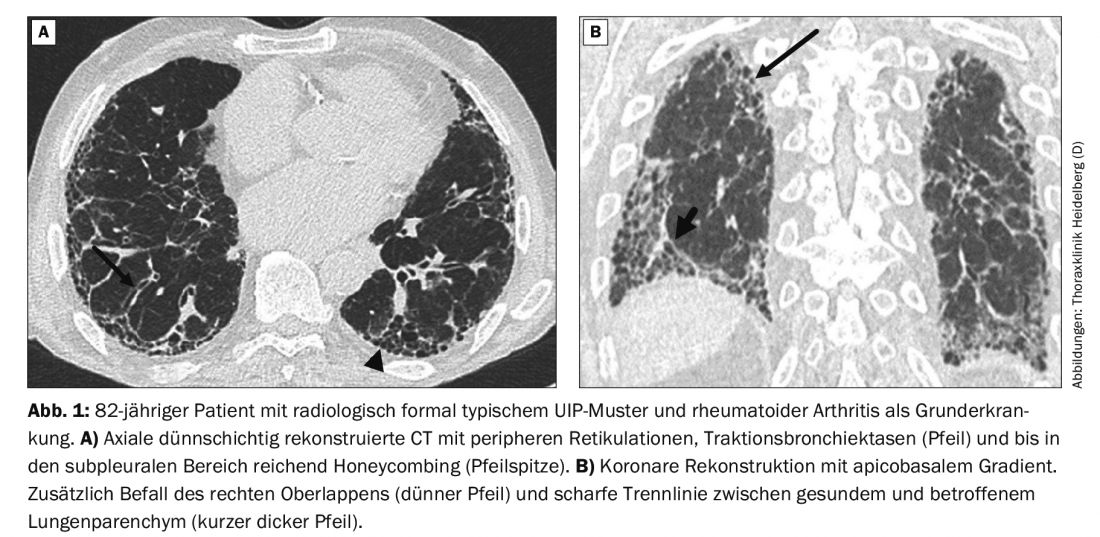
Um paciente de 82 anos apresenta-se com dispneia de esforço que tem sido progressiva durante 3-4 semanas. Há uma tosse sem expectoração. A exposição a substâncias perigosas não ocorreu. A história da família não tem nada a ver com isso. Até há 45 anos atrás, houve abuso ocasional de nicotina com um acumulado de 4-5 anos de pacote.
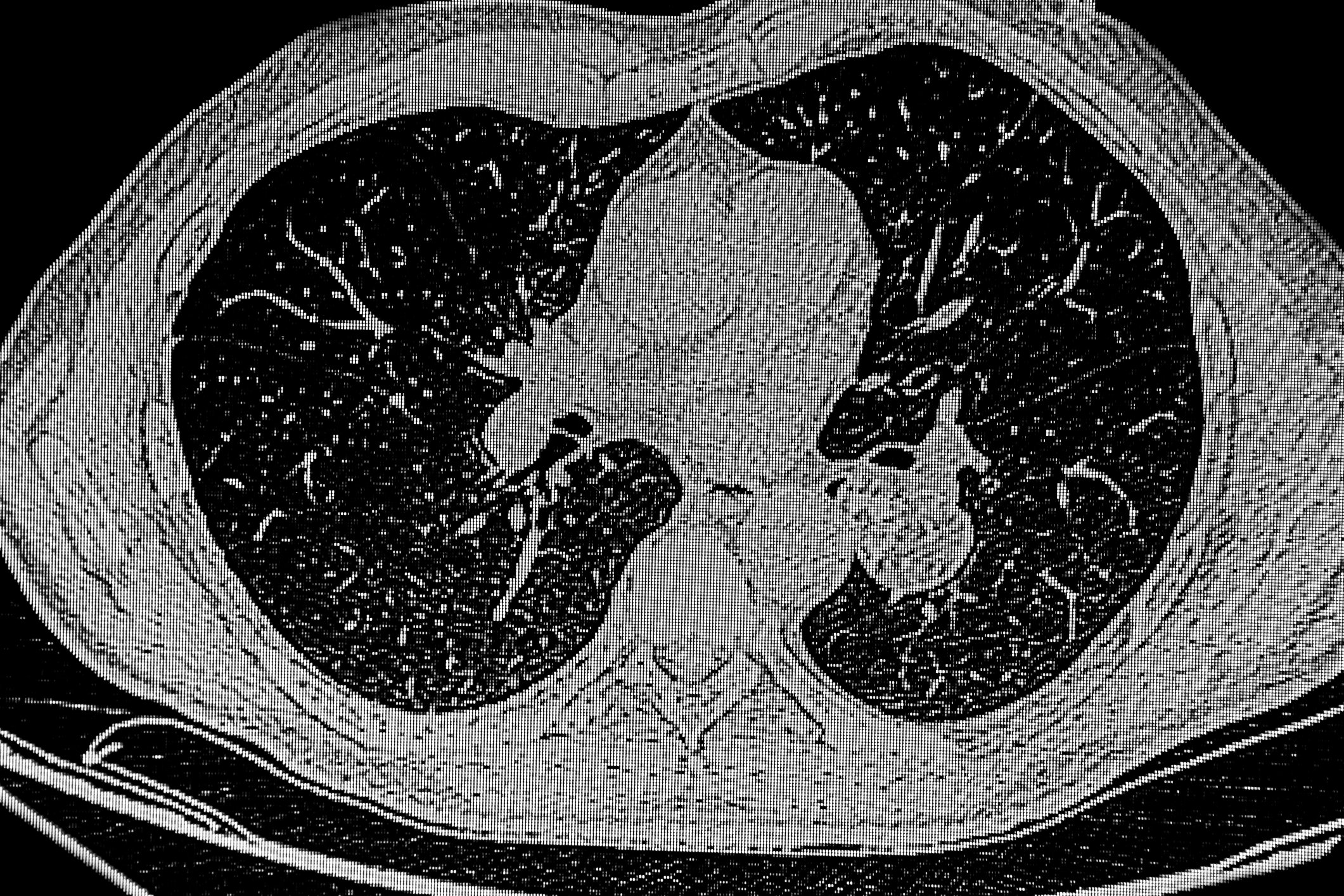
A digitalização CT nativa subsequente com reconstruções de camada fina mostra um padrão típico de UIP (Fig. 1A) . Durante a discussão interdisciplinar dos resultados, sabe-se que o paciente também sofre de artrite reumatóide (AR). Por conseguinte, o diagnóstico não é IPF mas ILD associado à RA.
Se um padrão ILD ocorrer no contexto de colagenose, o prognóstico para os pacientes é melhor do que no caso de IPF. Mesmo com um padrão UIP formalmente típico, provável ou indeterminado, em alguns casos há provas morfológicas adicionais no parênquima pulmonar de associação com a doença auto-imune [10]. No caso do paciente de 82 anos, trata-se do envolvimento adicional do lobo superior direito e da linha divisória oblíquo-horizontal aguda entre pulmão saudável e pulmão afectado (Fig. 1B). No entanto, estes sinais não são conclusivos e também podem ser encontrados em casos de IPF.
Os dados sobre a incidência de doença pulmonar intersticial (DPI) directamente causada pela AR variam, uma vez que os sintomas respiratórios aparecem frequentemente tardiamente devido à restrição do movimento. A DPI clinicamente significativa pode ocorrer em aproximadamente 7% dos pacientes de AR [11]. No entanto, as provas radiológicas de DPI (geralmente ainda assintomáticas) podem ser detectadas em tomografias computorizadas nativas reconstruídas em fatias finas em até 60% de todos os pacientes de AR [12]. O abuso da nicotina, que também pode datar de há vários anos, também aumenta o risco de colagenose associada à ILD com padrão UIP [13].
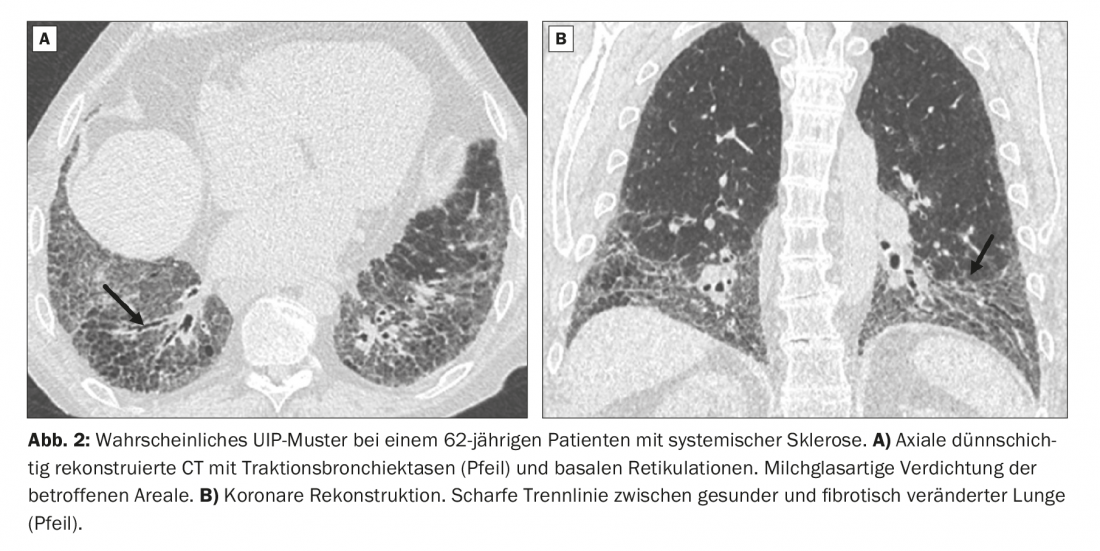
Categoria 2: provável padrão UIP
Um paciente de 62 anos apresenta-se com dispneia por esforço ligeiro. A esclerose sistémica já é conhecida. As imagens CT mostram um padrão estriado de bronquiectasia de tracção periférica e bronquiolectasia com ênfase subpleural e basal. Além disso, ligeiro aumento da densidade do vidro leitoso nas áreas afectadas (Fig. 2A).
CT-morfologicamente, a imagem corresponde formalmente a um provável padrão UIP e teria de ser classificada como tal sem o conhecimento da anamnese. Neste caso, no entanto, o diagnóstico é de DPI no contexto de esclerose sistémica. A linha divisória horizontal nítida entre pulmão saudável e pulmão afectado pode ser facilmente vista nas imagens de TC reformatadas coronárias (Fig. 2B).
Categoria 3: Padrão indeterminado com respeito a UIP ou CT não indicativo de nenhum dos diagnósticos.
Um paciente de 60 anos apresenta-se com dispneia. Até há 10 anos atrás, houve um abuso de nicotina com um total de 20 anos de embalagem. Não são conhecidas alergias, doenças auto-imunes ou exposição a substâncias perigosas. O CT de fatias finas mostrou um padrão indeterminado de UIP (Fig. 3). O exame histológico de uma biópsia cirúrgica subsequente do lobo inferior direito revelou um padrão UIP. O IPF foi diagnosticado. Um achado adicional foi um carcinoma brônquico de pequenas células.
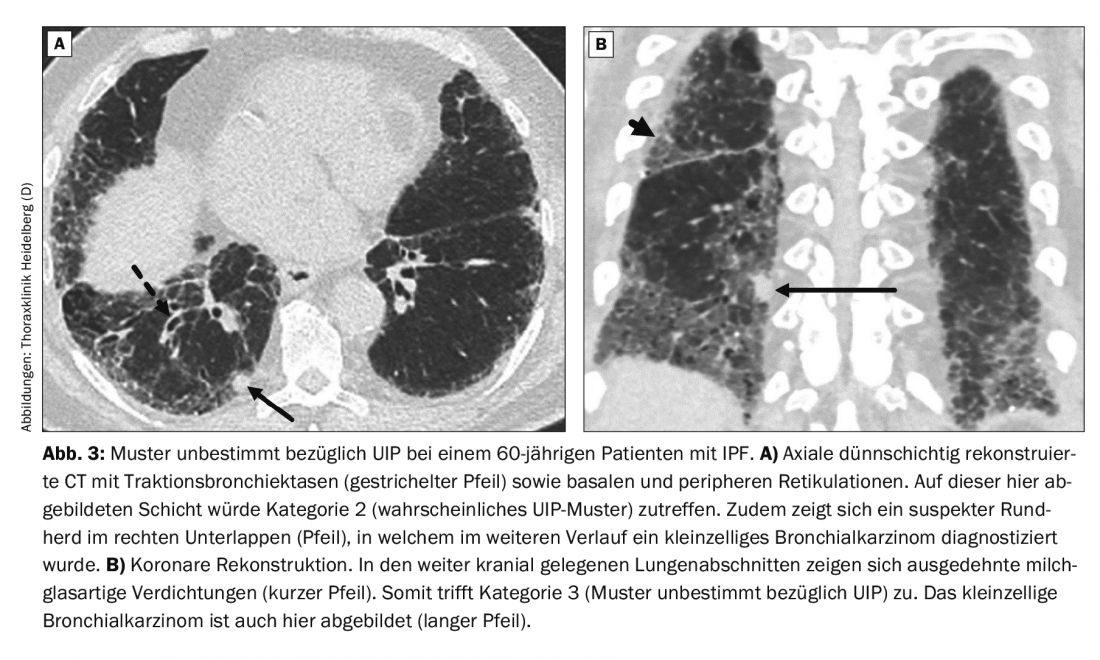
Categoria 4: provas morfológicas de um diagnóstico não-IPF
Um paciente de 71 anos com cardiomiopatia dilatada e 5 anos de terapia com amiodarona apresenta-se com dispneia de aumento lento. A pletismografia corporal mostrou uma doença pulmonar moderadamente restritiva. O padrão no CT de fatias finas não é compatível com um padrão UIP (Fig. 4). Inconsistentes com um padrão UIP são, por exemplo, o recesso da periferia subpleural e as elevações significativas da densidade do vidro leitoso, mesmo fora das áreas afectadas pela fibrose. Existe uma excepção: na pneumonia intersticial induzida por agentes patogénicos ou na exacerbação aguda da IPF, os infiltrados densos de vidro leitoso planos em combinação com alterações fibróticas pré-existentes são a imagem típica [14].
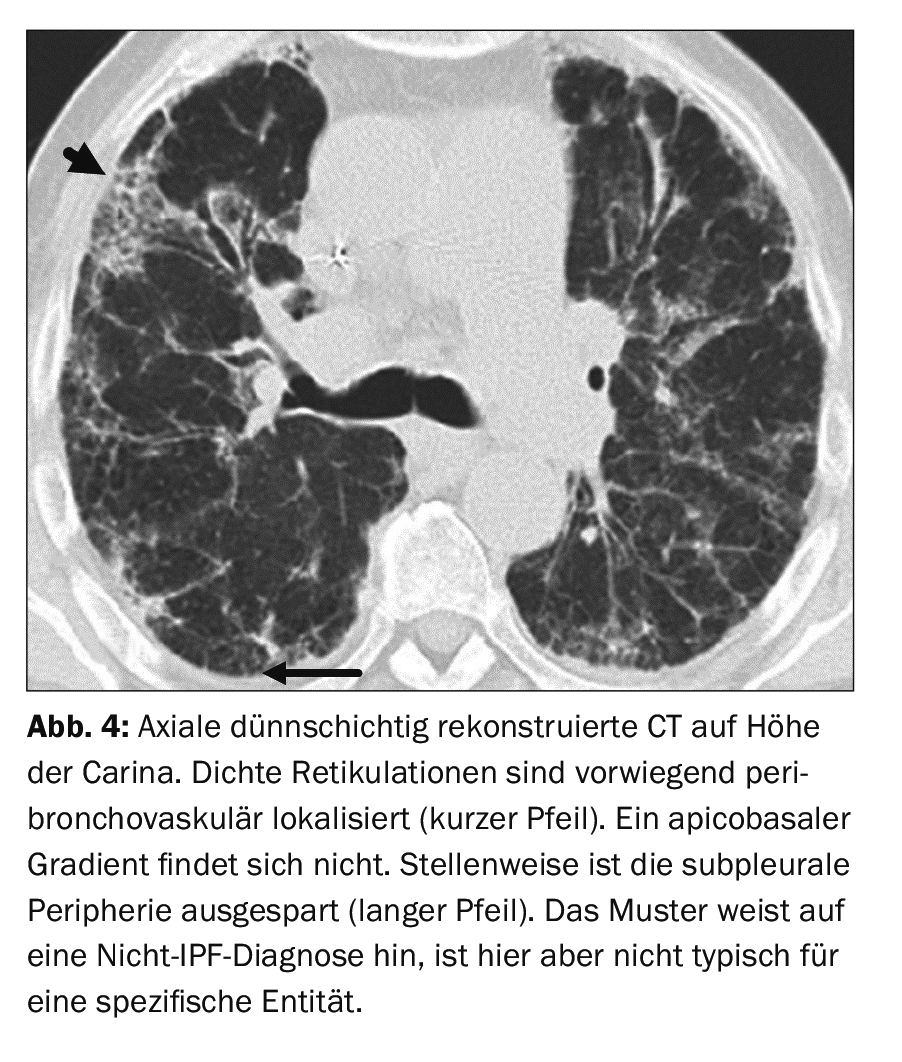
A consolidação pode ser uma indicação de cirurgia. A distribuição predominantemente peribrônquica, perilinfática ou exclusivamente nos segmentos apicais ou do pulmão médio também indica um diagnóstico não-IPF. Um padrão de mosaico claro, especialmente com três níveis de densidade e/ou micronódulos diferentes são indicações de CEA (pneumonia de hipersensibilidade, correspondente ao termo pneumonia de hipersensibilidade). A propósito, uma nova directriz para o diagnóstico das CEA foi também publicada em 2020 [6]. No caso do doente da Figura 4 , foi diagnosticada a DPI devido à toxicidade das drogas. Esta doença não tem um padrão específico ou patognomónico.
Mensagens Take-Home
- As chamadas doenças pulmonares intersticiais idiopáticas têm padrões característicos nas imagens de TC.
- O padrão de ouro para o diagnóstico da doença pulmonar intersticial é o consenso interdisciplinar (medicina interna, radiologia, patologia).
- O padrão UIP está dividido em 4 categorias.
- A classificação radiológica do padrão UIP, em combinação com dados clínicos, tem um impacto directo na decisão a favor ou contra uma biópsia pulmonar adicional e na decisão de tratamento.
Literatura:
- Capobianco J, et al: Manifestações Torácicas de Doenças Vasculares de Colagénio. RadioGraphics 2012; 32: 33-50.
- Travis WD, et al: Uma declaração oficial da American Thoracic Society/European Respiratory Society: Actualização da classificação internacional multidisciplinar das pneumonias intersticiais idiopáticas. Am J Respir Crit Care Med 2013; 188: 733-748.
- Lynch DA, et al: Diagnostic criteria for idiopathic pulmonary fibrosis: a Fleischner Society white paper. The Lancet Respiratory medicine 2018; 6: 138-153.
- Behr J: Diagnóstico e opções de tratamento para fibrose pulmonar idiopática. Dtsch Arztebl International 2013; 110: 875-881.
- Raghu G, et al: Prednisone, Azathioprine, e N-Acetylcysteine para Fibrose Pulmonar. N Engl J Med 2012; 366: 1968-1977.
- Raghu G, et al: Diagnóstico da Pneumonite de Hipersensibilidade em Adultos. Uma Directriz Oficial de Prática Clínica ATS/JRS/ALAT. Am J Respir Criteria Care Med 2020; 202: e36-e69
- Brownell R, et al: A utilização de probabilidades prévias aumenta o valor da CT de alta resolução no diagnóstico da pneumonia intersticial habitual. Tórax 2017; 72: 424-429.
- Tcherakian C, et al: Progressão da fibrose pulmonar idiopática: lições de doença assimétrica. Tórax 2011; 66: 226-231.
- Raghu G, et al: Diagnóstico da Fibrose Pulmonar Idiopática. Uma Directriz Oficial de Prática Clínica ATS/ERS/JRS/ALAT. American journal of respiratory and critical care medicine 2018; 198: e44-e68.
- Chung JH, et al: CT Features of the Usual Interstitial Pneumonia Pattern: Differentiating Connective Tissue Disease – Doença Pulmonar Intersticial Associada à Fibrose Pulmonar Idiopática. American Journal of Roentgenology 2017; 210: 307-313.
- Turesson C, et al: Manifestações de doenças extra-articulares na artrite reumatóide: tendências de incidência e factores de risco ao longo de 46 anos. Anais das doenças reumáticas 2003; 62: 722-727.
- Brown KK: Doença pulmonar reumatóide. Actas da American Thoracic Society 2007; 4: 443-448.
- Kumar A, et al: Current Concepts in Pathogenesis, Diagnosis, and Management of Smoking-Related Interstitial Lung Diseases. Peito 2018; 154: 394-408.
- Akira M, et al: Resultados da tomografia computorizada na exacerbação aguda da fibrose pulmonar idiopática. Am J Respir Crit Care Med 2008; 178: 372-378.
InFo PNEUMOLOGIA & ALERGOLOGIA 2021; 3(3): 6-10