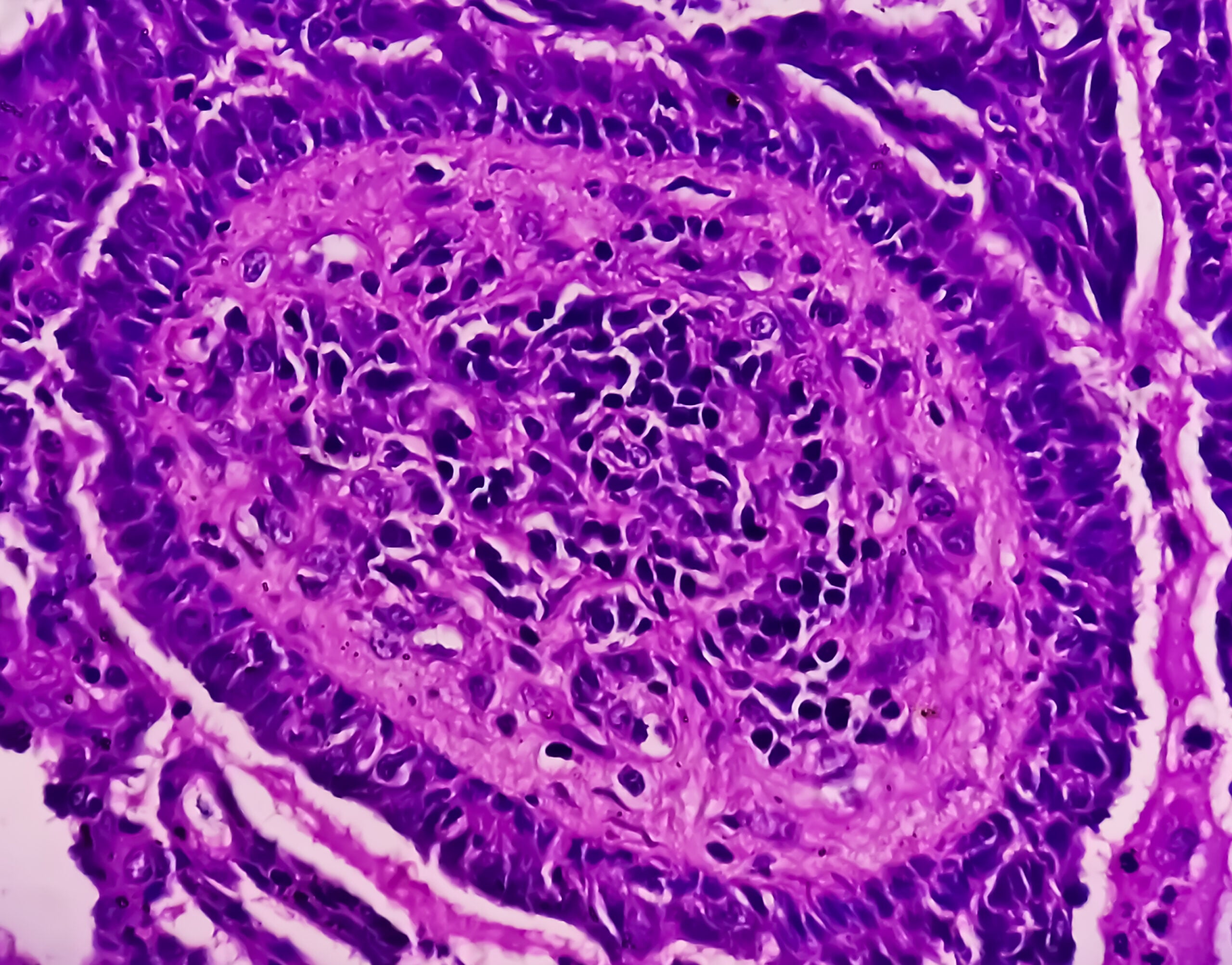
No mieloma múltiplo, a clínica que conduz ao diagnóstico é derivada da lesão do órgão final. Os critérios de diagnóstico foram actualizados pela última vez em 2014. Com diagnósticos bem estabelecidos e amplamente disponíveis, a doença raramente causa hoje em dia dificuldades de diagnóstico.
Com uma incidência anual de aproximadamente 5-6-6/100.000 e uma quota de 10%, o mieloma múltiplo é uma das neoplasias hematológicas mais comuns. Com uma idade média de 65-70 anos, as pessoas mais velhas são predominantemente afectadas, e os homens são afectados mais frequentemente do que as mulheres com uma proporção de 1,5:1 [1].
Patogénese
A transformação neoplásica de uma célula B do centro germinal em diferenciação para um plasmócito produtor de imunoglobulina é o evento iniciador da doença para um espectro de doenças de plasmócitos que se manifesta como gamopatia monoclonal de significado desconhecido (MGUS), mieloma de cheiro (SM), mieloma múltiplo (MM) ou leucemia de plasmócitos (PCL), dependendo da actividade da doença e da manifestação clínica [2].
A gamopatia monoclonal de significado pouco claro é uma lesão precursora clonal cuja incidência aumenta com a idade (ocorrendo em cerca de 3% dos >70 anos [3]), mas que apenas progride para o mieloma múltiplo em cerca de 1% dos casos por ano [4,5]. O risco exacto de progressão depende do tipo e da concentração da paraproteína, da relação da cadeia de luz livre, da percentagem de plasmócitos clonais na medula óssea e da imunoparesia [6,7].
Entretanto, a fase de mieloma assintomático chamado smouldering myeloma pode ser demarcada em aproximadamente 14% dos doentes, com uma taxa de progressão anual de 10% nos primeiros cinco anos após o diagnóstico inicial, seguida de 3% por ano nos próximos cinco anos e 1,5% nos anos seguintes [8,9]. Este é um estado de doença clinicamente definido entre MGUS e mieloma múltiplo que inclui uma população de doentes muito heterogénea, incluindo doentes com progressão de doença pré-maligna do tipo MGUS e doentes com mieloma múltiplo agressivo CRAB-negativo.
A leucemia de plasmócitos é a forma mais agressiva e leucémica de neoplasia de plasmócitos, com uma incidência de aproximadamente 4/10.000.000 [10] é comparativamente rara e pode desenvolver-se principalmente ou secundariamente a partir de um mieloma múltiplo pré-existente (1-4% de todos os doentes) [11]. Uma percentagem de plasmócitos de 20% ou uma concentração de 2000 plasmócitos/µl de sangue é necessária para o diagnóstico no hemograma diferencial microscópico.
O evento inicial na oncogénese das discrasias de plasmócitos ocorre numa fase de desenvolvimento das células B que se caracteriza per se por instabilidade genética devido a alterações na classe isotípica da molécula de imunoglobulina e hipermutação somática visando a maturação da afinidade [12].
Citogeneticamente, podem distinguir-se duas alterações principais do cariótipo, que no sentido de mutações primárias já estão presentes no início da oncogénese na fase de MGUS. Os cariótipos hiperdiplóides, observados em quase dois terços dos casos, caracterizam-se por trissomias em cromossomas com um número ímpar (3,5,7,9,11,15,19) e distinguem-se do chamado cariótipo não hiperdiplóide, que é frequentemente causado por translocações do locus da imunoglobulina de cadeia pesada (IgH) com oncogenes tais como FGFR-3 e MMSET (t[4;14]), MAF (t[14;16]), CCND1 (t[11;14]) ou é caracterizada por ganhos/perdas desequilibrados de 1q, 1p, 6q, 8p, 13q, 16q e 17p.
As alterações secundárias incluem mutações nas proteínas RAS (K/N-RAS), activando mutações em kinases como PI3K, AKT, BRAF, translocações com activação de factores de transcrição como MYC, e supressões ou inactivação de genes supressores de tumores como p53 e RB1 [13].
Ao mesmo tempo, o curso da doença caracteriza-se por uma crescente heterogeneidade clonal e instabilidade genómica [14], que é possivelmente aumentada pela intervenção quimioterapêutica (por exemplo, agentes alquilantes).
Uma vez estabelecido um clone de plasmócito maligno, os danos clínicos dos órgãos finais desenvolvem-se com o aumento da actividade da doença. Lesões ósseas osteolíticas devido ao aumento da reabsorção óssea são o resultado de metabolismo ósseo desregulado com aumento do osteoclasto e actividade osteoblasto suprimida, mediada pelo aumento da expressão RANKL (“activador receptor da NF kappa B ligand”), diminuição da expressão da osteoprotegerina [15] e um ambiente de citocinas osteoclasto-suportadoras (aumento da MIP-1 alfa, IL6, IL3, etc.). Outra consequência deste desequilíbrio é a libertação de cálcio da substância óssea com hipercalcemia do soro resultante e alterações na excitabilidade neuromuscular.
A anemia que frequentemente leva ao diagnóstico inicial e ao seu esclarecimento é o resultado do deslocamento de hematopoiese saudável por plasmócitos malignos. Contudo, esta explicação não vai suficientemente longe, porque em muitos casos observa-se anemia pronunciada que não pode ser explicada por uma baixa infiltração comprovada de plasmócitos. Aqui, alterações no microambiente da medula óssea como a activação do caminho de sinalização TGFβ parecem desempenhar um papel importante, levando a uma concentração reduzida de células progenitoras hematopoiéticas na medula óssea de doentes com mieloma [16].
Além da infiltração directa dos plasmócitos como elemento patogénico, a paraproteína monoclonal secretada pelos plasmócitos desempenha por vezes um papel decisivo na patogénese. Embora uma síndrome de hiperviscosidade causada por elevadas concentrações de paraproteína seja comparativamente rara e seja então encontrada principalmente nas doenças do mieloma com IgM ou paraproteína IgA, o que pode ser explicado pela sua estrutura molecular mais complexa com ocorrência como pentamer (IgM) ou dimer (IgA), o dano do órgão final causado pela cadeia de luz amilóide tóxica na patogénese da amiloidose AL, por outro lado, já ocorre com baixas concentrações de paraproteína. No mieloma de cadeia ligeira em particular, as cadeias de luz livre filtrável glomerular levam a danos tubulares e obstrução (a chamada nefropatia fundida) devido ao seu menor tamanho molecular em comparação com a molécula de imunoglobulina completa.
Clínica
Os sintomas clínicos que, em última análise, levam à consulta médica e ao diagnóstico são derivados dos danos dos órgãos finais relacionados com a mieloma descritos na sua patogénese. Dor óssea devido a osteólise relacionada com mieloma, uma fractura osteolítica patológica, desempenho reduzido na presença de anemia ou uma tendência para a infecção conduzem frequentemente a uma apresentação médica. O efeito nefrotóxico das cadeias ligeiras pode levar à insuficiência renal até à insuficiência renal com os correspondentes sintomas de uraemia. Menos frequentemente, o mieloma múltiplo manifesta-se com arritmias cardíacas, sonolência ou outros sintomas relacionados com a hipercalcemia. O efeito tóxico da amiloidose da cadeia de luz na amiloidose AL pode levar a uma sintomatologia clínica muito diversificada. A insuficiência cardíaca devida a depósito cardíaco é frequentemente encontrada aqui, juntamente com insuficiência renal, polineuropatia e outras.
Diagnósticos
Os critérios de diagnóstico do mieloma múltiplo foram actualizados pela última vez em 2014 pelo Grupo de Trabalho Internacional sobre Mieloma Múltiplo (IMWG) numa actualização consensual [17].
Os critérios CRAB estabelecidos (hipercalcemia, insuficiência renal, anemia, osteólise) foram complementados pelos chamados critérios SLiM (Tab. 1) . O contexto para a extensão dos critérios de diagnóstico é a observação de que no colectivo de doentes com mieloma latente que não necessitam de tratamento prévio, certos parâmetros da doença estão associados a uma elevada probabilidade de progressão (>80% no prazo de dois anos) estão associados ao mieloma múltiplo que requer tratamento e este grupo de doentes beneficia de uma intervenção terapêutica mais precoce [17,18].

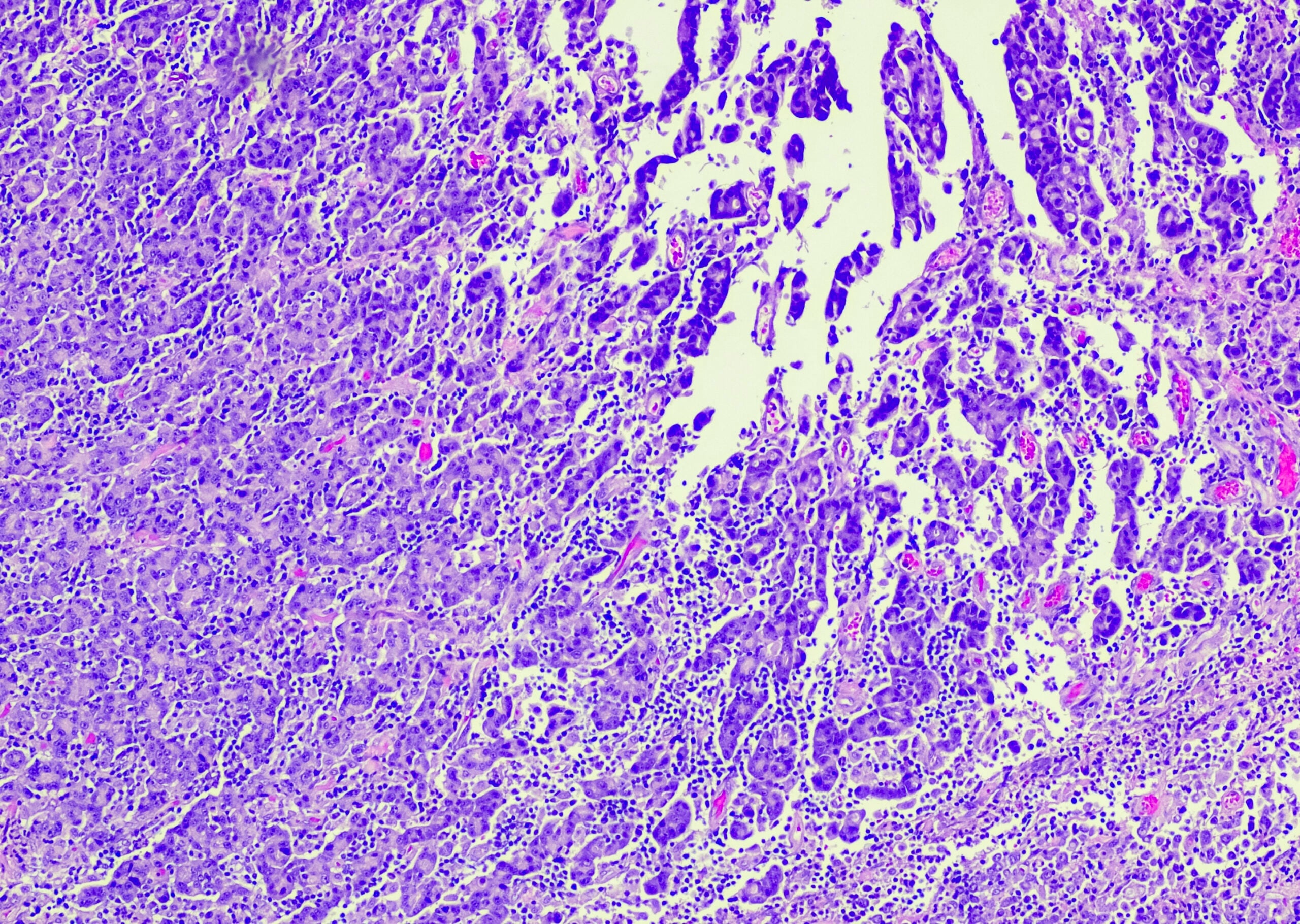
Assim, para o diagnóstico do mieloma múltiplo, é necessária a detecção de ≥10% de plasmócitos clonais na biópsia ou aspiração da medula óssea. (Fig.1) ou é necessária uma biopsia documentada do plasmacitoma ósseo ou extramedular, em conjunto com provas de um ou mais danos de órgãos finais ou biomarcadores de malignidade. (Tab.1). A prova da clonalidade dos plasmócitos é obtida através da detecção citométrica de fluxo de uma restrição de cadeia de luz citoplasmática. (Fig. 2) ou por coloração imuno-histoquímica das cadeias ligeiras numa biopsia representativa de punção de medula óssea.
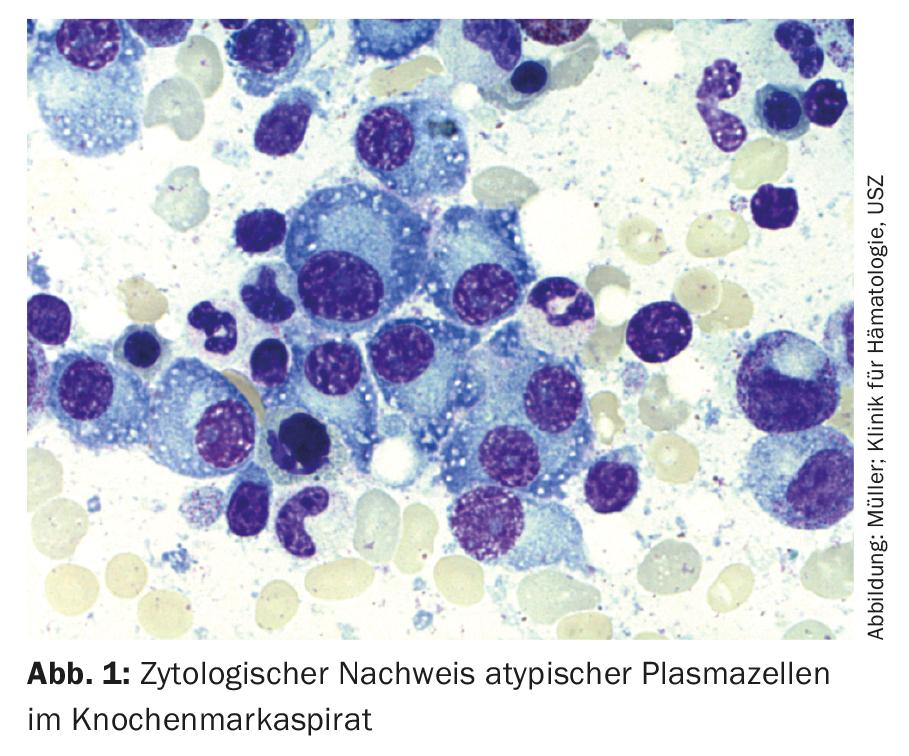
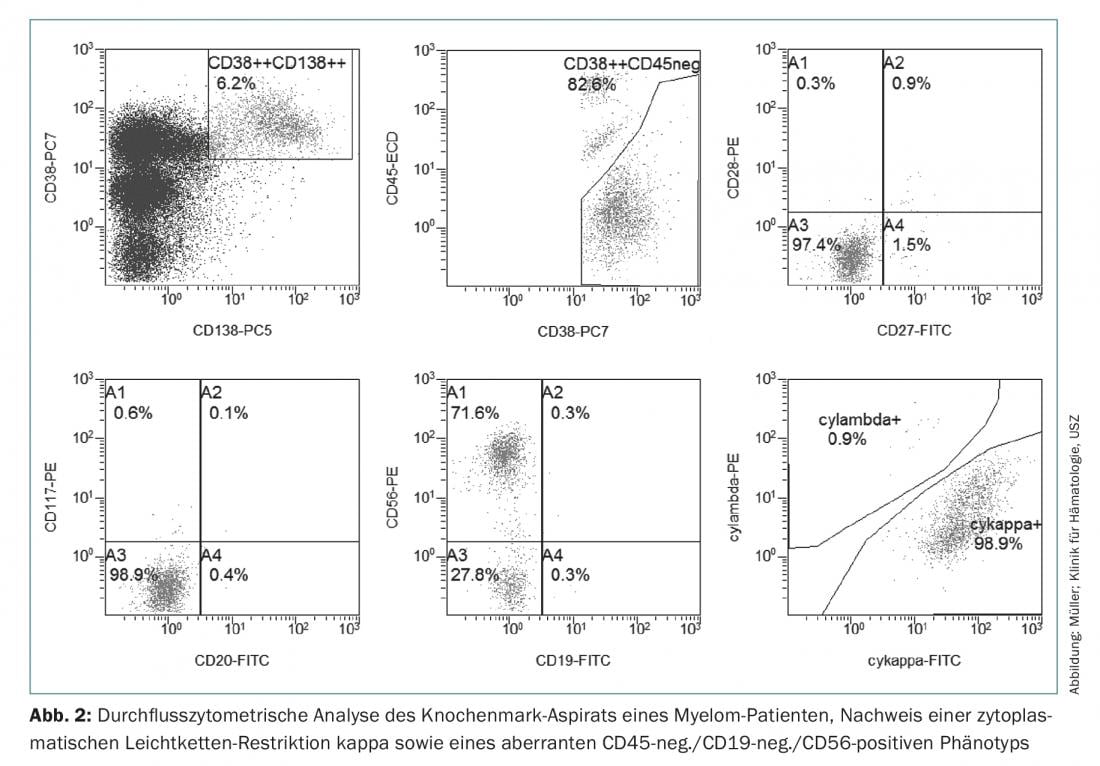
Imunofenotípicamente, as células do mieloma múltiplo podem ser distinguidas das saudáveis CD38++, CD138+, CD19+, CD45+ e CD56 – plasmócitos negativos por negatividade CD45 ou expressão reduzida, perda de CD19 e expressão de marcadores aberrantes como CD56, CD117 ou CD28, que podem ser utilizados no diagnóstico inicial, mas especialmente no diagnóstico do MRD, e por vezes têm também significado prognóstico [19].
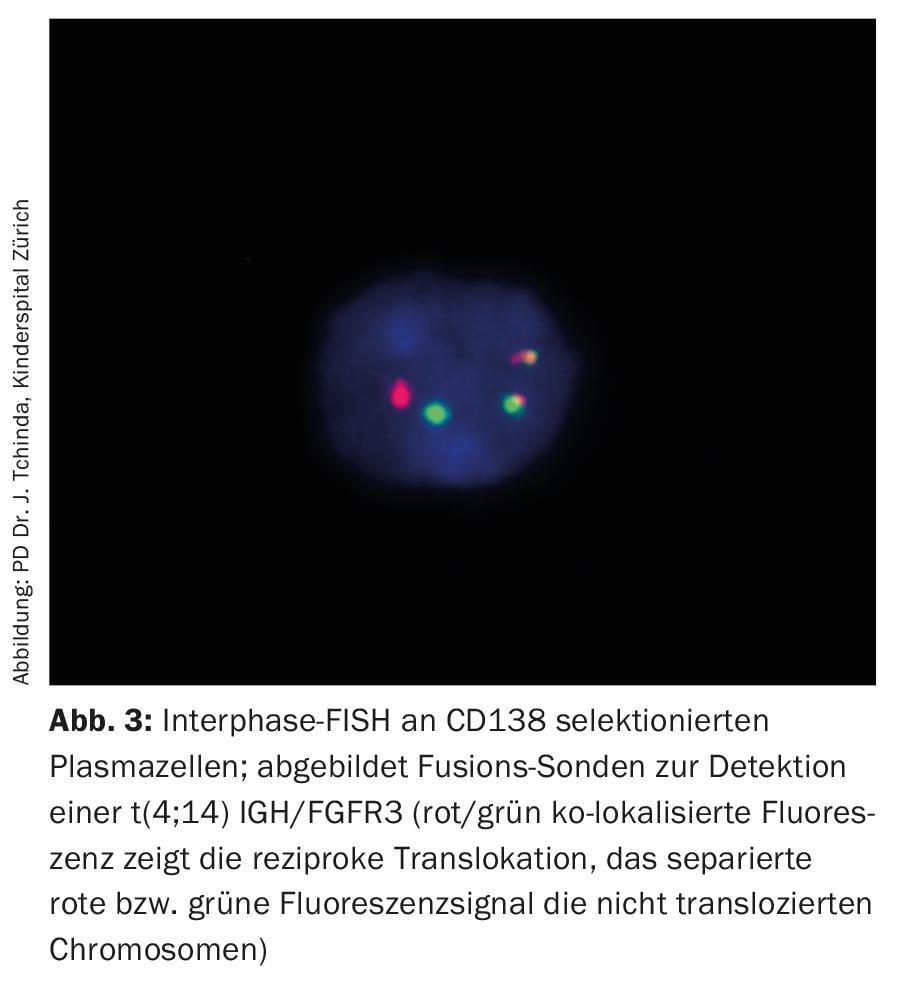
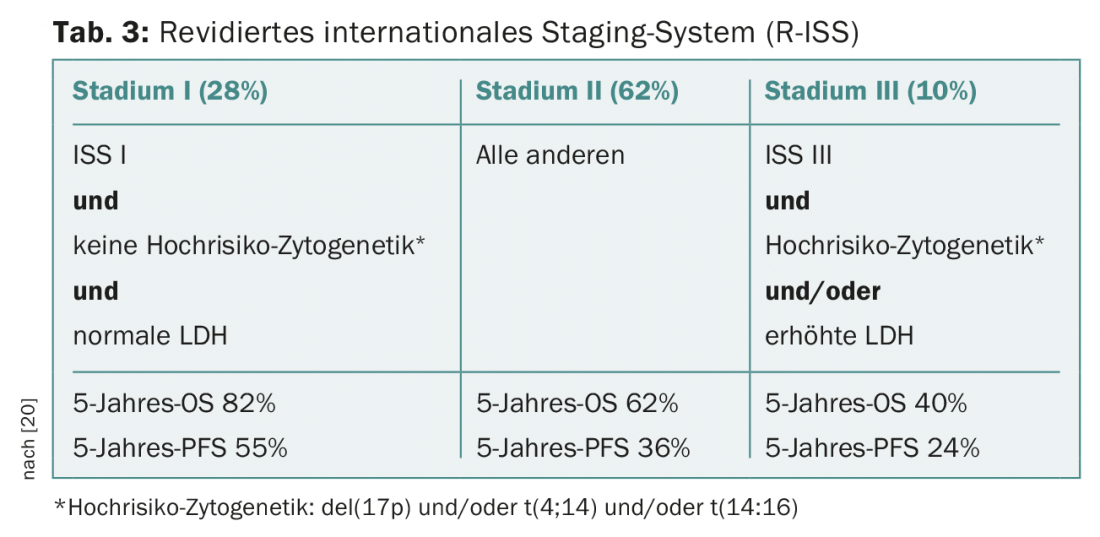
Enquanto a análise convencional do cariótipo das metáfases presas no mieloma múltiplo permanece geralmente pouco significativa devido ao baixo índice proliferativo e às difíceis condições de cultura celular, a hibridação fluorescente in situ dos núcleos interfásicos com sondas fluorescentes faz parte do trabalho padrão e, juntamente com os parâmetros de substituição estabelecidos da actividade da doença, tais como β2-microglobulina, albumina e lactato desidrogenase (LDH), permite a estratificação do risco a priori (Fig. 3, Tab. 3) [20].
O quadro 2 dá uma visão geral dos diagnósticos laboratoriais a serem realizados no diagnóstico inicial, bem como das imagens necessárias por meio de tomografia computorizada de baixa dose de corpo inteiro e, opcionalmente, ressonância magnética e PET-CT.
Com diagnósticos bem estabelecidos e amplamente disponíveis, o mieloma múltiplo raramente causa hoje em dia dificuldades de diagnóstico. Os desafios futuros residem antes numa subclassificação genética cada vez mais determinada da entidade da doença, com o objectivo de melhorar a estratificação do risco e o estabelecimento de marcadores preditivos para uma resposta terapêutica.
No contexto de opções de tratamento cada vez mais eficazes com melhor resposta e remissões mais profundas, a detecção da doença residual mínima (DRM) por meio da citometria de fluxo e sequenciação da próxima geração está também a desempenhar um papel cada vez mais importante na resposta e avaliação das remissões. Embora a importância prognóstica do MRD para a sobrevivência sem progressão (PFS) e sobrevivência global (OS) já tenha sido demonstrada [21], as decisões terapêuticas clínicas baseadas no MRD ainda não estão estabelecidas, mas são objecto de ensaios clínicos actuais.
Mensagens Take-Home
- A clínica que conduz à consulta médica e ao diagnóstico é derivada dos danos dos órgãos finais relacionados com a mieloma múltiplo.
- Os critérios de diagnóstico do mieloma múltiplo foram actualizados pela última vez em 2014, como parte de uma actualização consensual.
- Com diagnósticos bem estabelecidos e amplamente disponíveis, o mieloma múltiplo raramente causa hoje em dia dificuldades de diagnóstico.
- Pelo contrário, o desafio futuro reside numa subclassificação genética cada vez mais determinada da entidade da doença com o objectivo de melhorar a estratificação do risco e o estabelecimento de marcadores preditivos para uma resposta terapêutica.
- O significado prognóstico da doença residual mínima (DRM) para a sobrevivência sem progressão (PFS) e sobrevivência global (SO) foi demonstrado, e as decisões clínicas de tratamento baseadas na DRM são objecto de ensaios clínicos actuais.
Literatura:
- Rodriguez-Abreu D, Bordoni A, Zucca E: Epidemiologia de malignidades hematológicas. Annals of Oncology 2007; 18(Suppl 1): i3-i8.
- Bakkus MH, et al: Evidência de que os genes VDJ de cadeia pesada do mieloma múltiplo contêm mutações somáticas mas não mostram variação intraclonal. Sangue 1992; 80: 2326-2335.
- Kyle RA, et al: Prevalência da gamopatia monoclonal de significado indeterminado. N Engl J Med 2006; 354: 1362-1369.
- Landgren O, et al: A gamopatia monoclonal de significado indeterminado (MGUS) precede consistentemente o mieloma múltiplo: um estudo prospectivo. Sangue 2009; 113: 5412-5417.
- Weiss BM, et al: Uma gamopatia monoclonal precede o mieloma múltiplo na maioria dos doentes. Sangue 2009; 113: 5418-5422.
- Baldini L, et al.: Papel de diferentes variáveis hematológicas na definição do risco de transformação maligna na gamopatia monoclonal. Sangue 1996; 87: 912-918.
- Turesson I, et al: Gamopatia monoclonal de significado indeterminado e risco de malignidades linfóides e mielóide: 728 casos seguidos até 30 anos na Suécia. Sangue 2014; 123: 338-345.
- Kristinsson SY, Holmberg E, Blimark C: Tratamento para mieloma de alto risco de combustão lenta. N Engl J Med 2013; 369: 1762-1763.
- Kyle RA, et al: Curso clínico e prognóstico do mieloma múltiplo (assintomático) de combustão lenta. N Engl J Med 2007; 356: 2582-2590.
- Sant M, et al: Incidência de malignidades hematológicas na Europa por subtipo morfológico: resultados do projecto HAEMACARE. Sangue 2010; 116: 3724-3734.
- Tiedemann RE, et al.: aberrações genéticas e sobrevivência na leucemia de plasmócitos. Leucemia 2008; 22: 1044-1052.
- Seifert M, Scholtysik R, Küppers R: Origem e patogénese dos linfomas das células B. Métodos Mol Biol 2013; 971: 1-25.
- Kuehl WM, Bergsagel PL: mieloma múltiplo: evolução de eventos genéticos e interacções com o hospedeiro. Nat Rev Cancer 2002; 2: 175-187.
- Bolli N, et al.: Heterogeneidade da evolução genómica e perfis mutacionais no mieloma múltiplo. Nat Commun 2014; 5: 2997.
- Roodman GD: Mecanismos de metástase óssea. N Engl J Med 2004; 350: 1655-1664.
- Bruns I, et al.: Desregulação de células estaminais hematopoiéticas e progenitoras CD34(+) derivadas da medula óssea relacionada com mieloma múltiplo. Sangue 2012; 120: 2620-2630.
- Rajkumar SV, et al: International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol 2014; 15: e538-48.
- Mateos MV, et al: Lenalidomida mais dexametasona para mieloma múltiplo de alto risco de combustão lenta. N Engl J Med 2013; 369: 438-447.
- Mateo G, et al: Prognostic Value of Immunophenotyping in Multiple Myeloma: A Study by the PETHEMA/GEM Cooperative Study Groups on Patients Uniformly Treated With High-Dose Therapy. Journal of Clinical Oncology 2008; 26: 2737-2744.
- Palumbo A, et al: Revised International Staging System for Multiple Myeloma: A Report From International Myeloma Working Group. J Clin Oncol 2015 Set 10; 33(26): 2863-2869.
- Paiva B, van Dongen JJM, Orfao A: Novos critérios de avaliação da resposta: papel da doença residual mínima no mieloma múltiplo. Sangue 2015; 125: 3059-3068.
- Moreau P, et al: Mieloma múltiplo: Directrizes de Prática Clínica da OMPE para diagnóstico, tratamento e acompanhamento? Ann Oncol 2017 Jul 1; 28(suppl_4): iv52-iv61.
InFo ONCOLOGy & HEMATOLOGy 2017; 5(5): 7-10