A neoplasia mieloproliferativa das células estaminais hematopoiéticas é predominantemente causada por mutações somáticas do gene JAK2 e pela hematopoiese clonal autónoma resultante. O risco de mielofibrose ou transformação leucémica são prognosticamente relevantes.
A Policitemia vera (PV) é actualmente classificada pela Organização Mundial de Saúde (OMS) na categoria principal das neoplasias mieloproliferativas (NMP) [1,2]. Segundo os conhecimentos actuais, a PV é uma doença do tronco hematopoiético ou células progenitoras, causada predominantemente por mutações somáticas no gene JAK2, com a consequente mieloproliferação clonal [3]. Foram descritos grupos familiares raros, com mutações da linha germinal no receptor de eritropoietina (EPOR) [4]. A incidência da PV na Europa é de 0,4-2,8% da população por ano. A idade média de início é de 60-65 anos, e as mulheres e os homens são afectados em número aproximadamente igual [5,6]. O tempo médio de sobrevivência é de 14 anos nos doentes com PV mais velhos (>60 anos) e 24 anos nos com menos de 60 [7].
Clínica
No quadro sanguíneo, a doença manifesta-se por um aumento da produção de eritrócitos (eritrocitose) independente dos mecanismos de regulação fisiológica. Leucocitose e trombocitose devido ao aumento da megacariopoiese acompanham frequentemente a doença. No decurso da doença, alterações fibróticas progressivas no sentido da mielofibrose levam a uma chamada “fase gasta” com hematopoiese reduzida em até 10% de todos os doentes com FV [8]. A transformação leucémica é diagnosticada em até 5% dos casos de FV após 20 anos de progressão da doença [7,9]. Os factores de risco prognósticos para mielofibrose, transformação leucémica e sobrevivência global reduzida são a idade avançada do doente, leucocitose, trombocitose e um cariótipo anormal [10].
Além disso, a ocorrência de eventos tromboembólicos venosos e arteriais em 20-50% de todos os pacientes com FV é de importância prognóstica decisiva. Patofisiologicamente, estes baseiam-se no aumento da viscosidade sanguínea e nas alterações reológicas resultantes, bem como em estímulos inflamatórios, procoagulantes e microvasculares [11,12]. Distinguem-se dois grupos de risco de trombose recorrente: Alto risco em doentes >60 anos e história positiva de eventos trombóticos e de um grupo de baixo risco, na ausência de ambos os factores [13]. A tensão arterial elevada é um factor de risco independente para a ocorrência de trombos arteriais.
Clinicamente, a PV manifesta-se principalmente por fadiga (até 84,9%) [14] e prurido (em cerca de 40%) [15]. Outros sintomas clínicos incluem pele facial avermelhada, pele vermelha-azulada e membranas mucosas, pressão na cabeça, dor de cabeça e hipertensão. As perturbações microcirculatórias conduzem frequentemente a sintomas clínicos característicos (por exemplo, perturbações visuais, paraestesias, eritromelalgia, queixas pectanginais) [16]. A trombose venosa abdominal e a trombose da veia sinusal são outras complicações com consequências graves e muitas vezes fatais [17,18]. Paradoxalmente, em casos raros com trombocitose extrema (>1000×109/L) há também uma tendência crescente para a hemorragia. Isto é causado por uma redução do factor Von Willebrand (VWF) no sentido de uma síndrome de consumo adquirido. Pathobiologicamente, tanto o aumento da actividade do ADAMTS13 como o aumento da sensibilidade às plaquetas e a consequente activação inadequada das plaquetas desempenham um papel significativo [19,20]. A esplenomegalia encontra-se em cerca de um terço de todas as pessoas afectadas e pode ser associada à dor e ao enfarte.
Patobiologia da PV
A PV é caracterizada pela mieloproliferação clonal devido a alterações patológicas nas células estaminais hematológicas [3]. Quase todos os doentes com PV têm uma mutação de Janus kinase 2 (JAK2), que se localiza no gene locus 9p24, sendo que 96% de todos os casos têm uma mutação somática activadora no exon 14 (JAK2V617F) [21] e menos frequentemente (cerca de 3%) no exon 12 [22]. Até agora, não foram demonstradas diferenças significativas no curso clínico, dependendo da respectiva mutação [23].
Em condições fisiológicas, a ligação da eritropoietina (EPO) ao receptor EPO (EPOR) leva a uma alteração conformacional (homodimerização do receptor) com a resultante auto-fosforilação e activação de JAK. Moléculas JAK activadas de fosforilato e subsequentemente activam moléculas STAT (transdutor de sinal e activador de transcrição), as quais, após translocação para o núcleo celular, activam sinais de crescimento hematopoiético como factores de transcrição [24]. A activação da mutação JAK2V617F resulta na activação constitucional independente do EPO JAK/STAT e na proliferação incontrolada de células (Fig. 1) [25]. Outras alterações genéticas como a desregulação do apotosinibidor Bcl-x ou o factor de transcrição NF-E2 foram descritas, mas parecem ser consequências secundárias de uma mutação JAK2 [26].
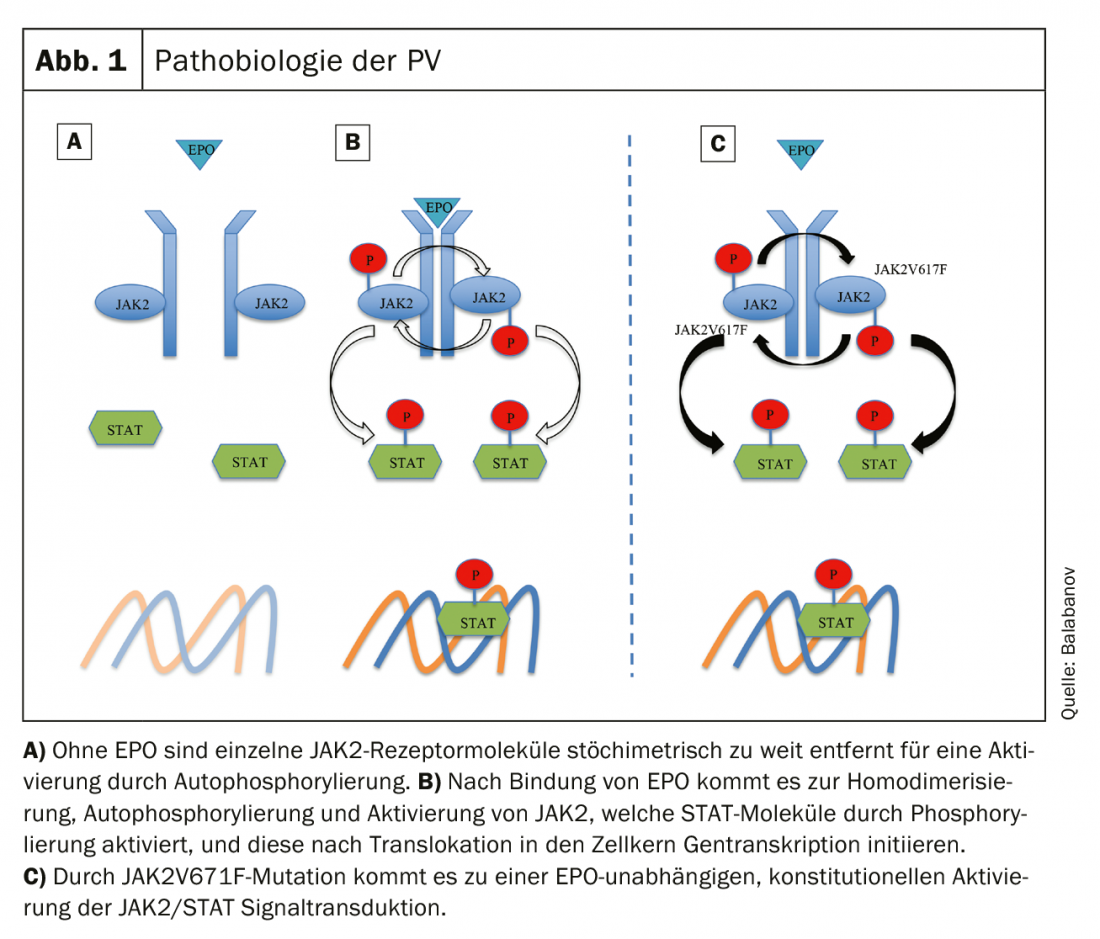
As mutações JAK2V617F são mais comuns em doentes idosos e estão associadas à panmielocitose, mielofibrose e sintomas clínicos de prurido [22,23]. Uma correlação com o risco global de sobrevivência ou de transformação não pôde ser provada [3]. A frequência dos alelos mutantes também não parece influenciar a sobrevivência ou a frequência da transformação leucémica. Num estudo realizado na Mayo Clinic, a sequenciação profunda orientada mostrou que mais de metade (53%) dos doentes examinados tinham mutações adicionais para além de JAK2 [28]. As mutações mais frequentes foram detectadas em TET2 (22%), ASXL1 (12%), SH2B3 (9%) (outras mutações foram encontradas em SRSF2, IDH2, TP53). Especialmente as mutações em ASXL1, SRSF2 e IHD2 mostraram um efeito prognóstico independente de outros preditores (mediana de sobrevivência 7,7 anos com vs. 16,9 anos sem mutações). Um cariótipo anormal é encontrado em 15% dos doentes no diagnóstico inicial, sendo as alterações citogénicas mais comuns detectadas -Y, +8, +9, del(20q) e 1q+ [27].
Diagnósticos
Um historial médico detalhado e um exame físico constituem a base do diagnóstico. O questionário Myeloproliferative Neoplasm Symptom Assessment Form (MPN-SAF) fornece uma avaliação estruturada e completa dos sintomas relevantes [29]. Em particular, devido à relevância do prognóstico e do tratamento, deve ser dada atenção aos eventos trombóticos na história, aos estigmas de trombose, à hipertensão e à idade do paciente. A ultra-sonografia abdominal é recomendada para avaliar a esplenomegalia.
Para além de um hemograma diferencial e do estado de coagulação, a concentração de EPO deve ser determinada em laboratório. O diagnóstico final só pode ser feito com base numa biópsia de medula óssea.
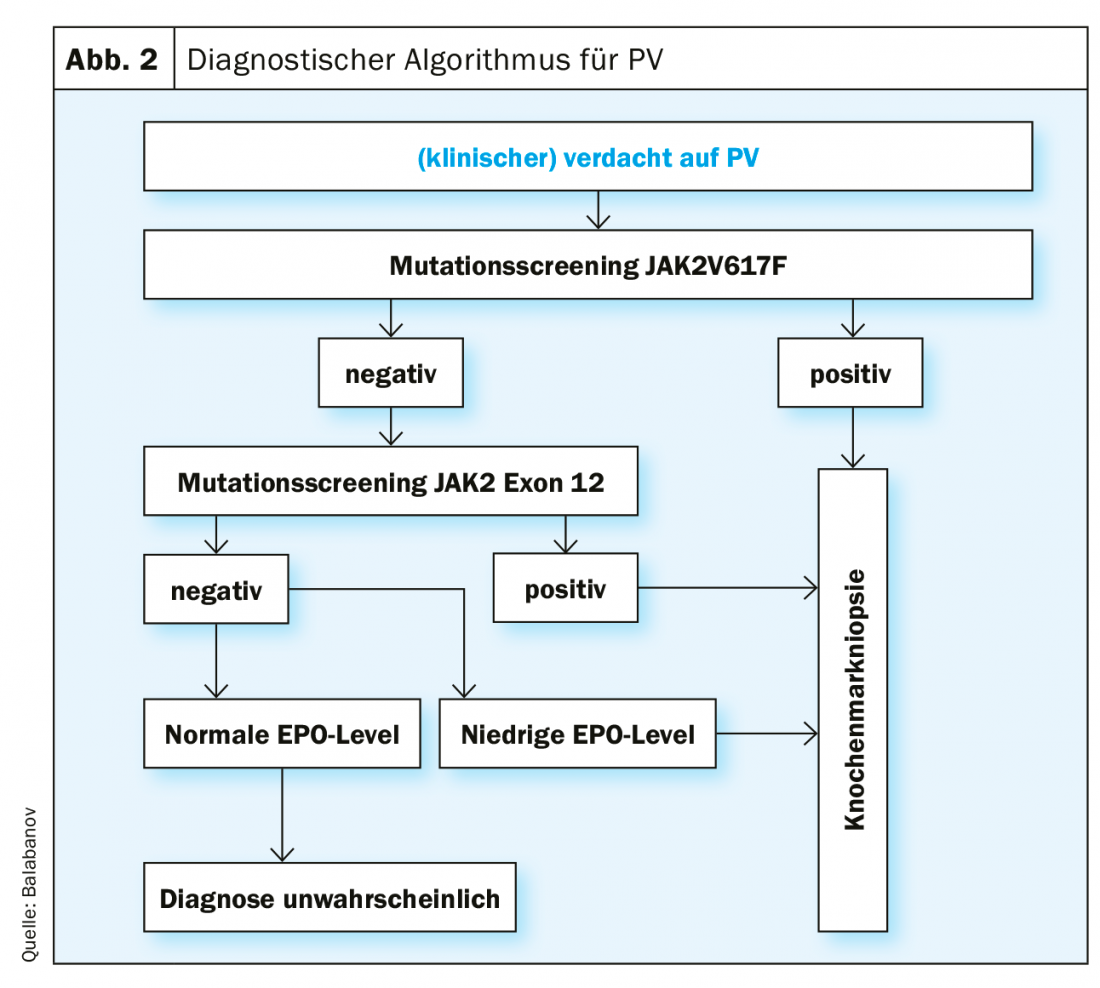
Os critérios de diagnóstico de acordo com a classificação actual (2016) da OMS são apresentados no Quadro 1. Para fazer um diagnóstico de PV, todos os critérios principais ou dois critérios principais e o critério menor devem ser cumpridos. A detecção de uma mutação JAK2V617F é extremamente fiável, com uma sensibilidade de teste de 97% e uma especificidade de quase 100%. Se o estado da mutação JAK2V617F for negativo mas o nível sérico EPO for baixo, é indicada uma análise de mutação adicional no exon 12 do gene JAK2. Devido à relevância prognóstica tanto para a sobrevivência global como para a sobrevivência sem leucemia, deve ser efectuada uma determinação do cariótipo, se necessário. Além disso, uma análise de mutação por meio de Next Generation Sequencing (NGS) é útil, visto que as mutações em ASXL1, SRSF2 e IDH2 são de relevância prognóstica. No entanto, a NGS ainda não é uma investigação padrão para cada paciente com PV no momento do diagnóstico. Contudo, um estudo de referência recentemente publicado demonstrou que a integração abrangente de variáveis genómicas e clínicas permite uma estratificação de risco personalizada com implicações terapêuticas [30] (https://cancer.sanger.ac.uk/mpn-multistage).

A Figura 2 mostra um algoritmo para o procedimento diagnóstico de suspeita de VP, com base nos actuais critérios de diagnóstico da OMS. Os testes funcionais especiais do VWF são indicados para esclarecer uma tendência crescente de hemorragia (por exemplo, actividade do co-factor ristocitina).
Terapia
Com uma taxa de sobrevivência de 10 anos superior a 75%, e um risco relativamente baixo de mielofibrose (<10%) e transformação maligna no sentido de transformação leucémica (<5%) [8], o principal objectivo do tratamento da PV é prevenir complicações trombo-hemorrágicas e aliviar os sintomas acima descritos.
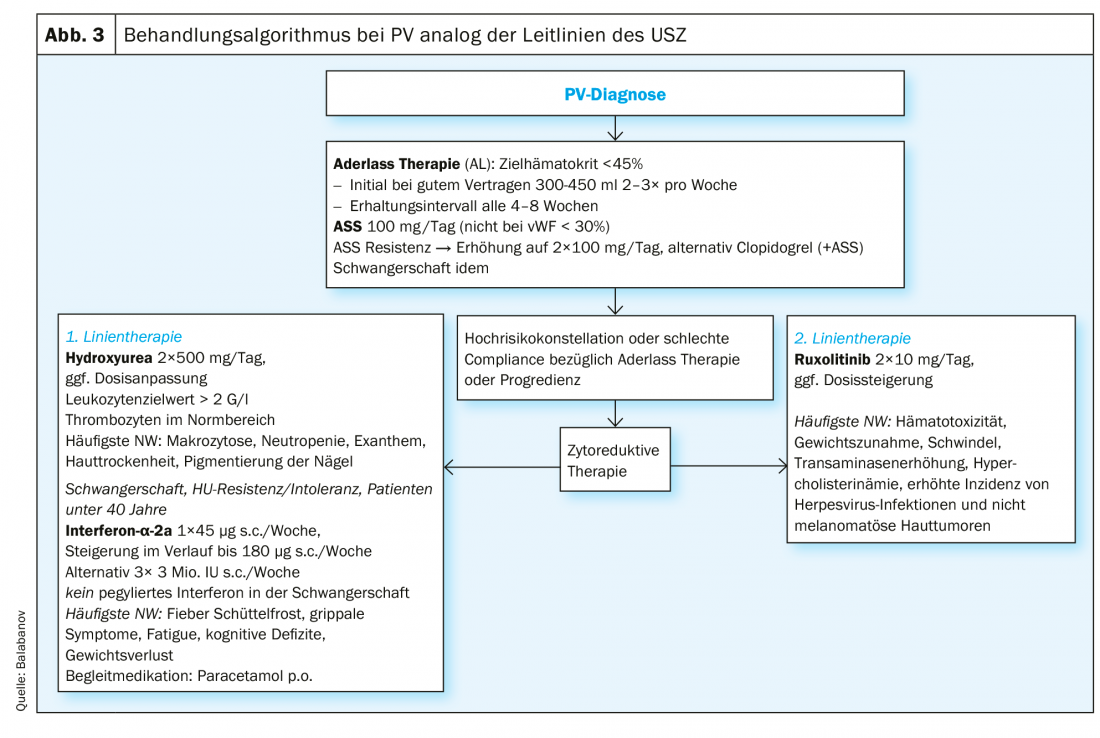
Pacientes de baixo risco (idade <60 anos, história negativa de eventos trombóticos): Recomendamos uma terapia de flebotomia consistente, visando um hematócrito alvo <45%, uma vez que este valor alvo é superior a valores superiores (45-50%) em termos de sobrevivência livre de eventos [31]. Um efeito positivo nos sintomas da doença, por outro lado, ainda não foi documentado [34]. Além disso, a terapia antiagregante com aspirina (ASA) é recomendada para prevenir trombos venosos e arteriais [31–33]. O tratamento com ASA parece ter uma influência positiva nos sintomas associados, reduzindo os distúrbios microvasculares [35]. No caso de sintomas resistentes à aspirina, pode ser considerada a administração duas vezes por dia ou alternativamente o uso de clopidogrel sozinho ou em combinação com ASA [36, 37]. Se necessário, é indicado um teste de função plaquetária (agregometria plaquetária, PFA-100) para distinguir defeitos congénitos de causas iatrogénicas [38]. Aconselha-se cautela no uso de aspirina, tendo em conta a possível tendência de aumento da hemorragia em doentes com trombocitose extrema concomitante (>1000×109/L).
Pacientes de alto risco (idade >60 anos, história positiva de eventos trombóticos): Para além das medidas terapêuticas acima mencionadas, recomendamos o tratamento citoreducativo neste grupo de doentes, com o objectivo principal de normalizar o hematócrito. No entanto, a redução da trombocitose e leucocitose que frequentemente acompanha a trombocitose e a leucocitose também deve ser visada. As recomendações actuais para substâncias para terapia de primeira linha de acordo com a LeukemiaNET (ENL) europeia são a hidroxiureia e o interferão α (INFα) (forma peguilada) [39]. O fracasso do tratamento sob hidroxiureia (tab. 2) no sentido de resistência ou intolerância ocorre em cerca de 24% dos doentes [7]. O Busulfan tem sido historicamente utilizado com bom sucesso na terapia de segunda linha. Actualmente, a sua utilização é essencialmente obsoleta devido à suspeita de um potencial leucemogénico, pelo que não estão disponíveis dados sólidos no que diz respeito ao aumento da ocorrência de leucemias sob o busulfan [40].

O inibidor JAK2 ruxolitinibe é aprovado como terapia de segunda linha para PV e mostrou resultados positivos em termos de mieloproliferação, necessidade de flebotomia e sintomas associados à PV, como fadiga e prurido com boa tolerância [41–44]. Para o prurido refractário, foi descrito o uso de SSRIs [45], INFα [46] e fototerapia [47]. Um resumo do algoritmo de tratamento do Hospital Universitário de Zurique (USZ) pode ser encontrado na Figura 3. O transplante alogénico de medula óssea ou o transplante de células estaminais do sangue periférico deve ser considerado como uma abordagem curativa apenas em casos excepcionais em pacientes mais jovens com um curso rico em complicações ou uma progressão da doença que não pode ser controlada por outras medidas [48]. Devido ao aumento do risco cardiovascular, todos os pacientes devem ser encorajados a reduzir o risco cardiovascular em termos de prevenção primária (exercício, controlo de peso, dieta).
Gravidez: a PV não é uma contra-indicação para a gravidez, mas estas são sempre consideradas gravidezes de alto risco com uma taxa aumentada de abortos espontâneos em comparação com a população normal [49]. Em princípio, recomendamos cuidados num centro especializado com conhecimentos hematológicos adequados. As recomendações relativas à flebotomia e à terapia anti-agregante são essencialmente as mesmas que as acima mencionadas. A Aspirina parece reduzir a taxa de abortos pontuais e complicações na gravidez [50]. Devido à falta de provas de teratogenicidade ou influência sobre a taxa de natalidade [51] com IFNα é geralmente possível e especialmente recomendada em gravidezes de alto risco [52].
Mensagens Take-Home
- A policitemia vera é uma neoplasia mieloproliferativa de células estaminais hematopoiéticas predominantemente desencadeada por mutações somáticas do gene JAK2 e pela hematopoiese clonal autónoma resultante.
- Clinicamente, os sintomas e complicações mirco- e macrovasculares no contexto da eritrocitose e frequentes trombocitose que a acompanham são proeminentes. De maior relevância prognóstica é o risco de mielofibrose e transformação leucémica.
- Para além dos parâmetros clínicos e laboratoriais, o algoritmo de diagnóstico envolve principalmente a detecção de uma mutação JAK2 na medula óssea, com outras aberrações genéticas a tornarem-se cada vez mais importantes.
- Terapêuticamente, os doentes são divididos em grupos de risco. Enquanto que a flebotomia e os inibidores de agregação plaquetária são utilizados independentemente do risco, a terapia citoreducativa é indicada principalmente em doentes de alto risco.
- As gravidezes em doentes com FV são sempre consideradas gravidezes de alto risco e devem ser geridas de forma multidisciplinar em centros especializados.
Literatura:
- Arber DA, et al: A revisão de 2016 da classificação da Organização Mundial de Saúde das neoplasias mielóides e da leucemia aguda. Sangue, 2016. 127(20): 2391-2405.
- Barbui T, et al: A revisão de 2016 da classificação da OMS das neoplasias mieloproliferativas: Avanços clínicos e moleculares. Blood Rev, 2016. 30(6): 453-459.
- Jamieson CH, et al.: A mutação JAK2 V617F ocorre em células estaminais hematopoiéticas em policitemia vera e predispõe para a diferenciação eritróide. Proc. Natl Acad Sci U S A, 2006. 103(16): 6224-6229.
- Bento C, et al: Genetic basis of congenital erythrocytosis: mutation update and online databases. Hum Mutat, 2014. 35(1): 15-26.
- Policitemia vera: a história natural de 1213 doentes seguiu-se durante 20 anos. Gruppo Italiano Studio Policitemia. Ann Intern Med, 1995. 123(9): 656-664.
- Moulard O, et al: Epidemiologia da mielofibrose, trombocitémia essencial, e policitemia vera na União Europeia. Eur J Haematol, 2014. 92(4): 289-297.
- Tefferi A, et al.: Sobrevivência a longo prazo e transformação por explosão em trombocitémia essencial anotada molecularmente, policitemia vera, e mielofibrose. Sangue, 2014. 124(16): 2507-2513; quiz 2615.
- Crisa E, et al.: Um estudo retrospectivo em 226 doentes com policitemia vera: impacto do valor médio do hematócrito nos resultados clínicos e melhoria da sobrevivência com profilaxia antitrombótica e medicamentos não alquilantes. Ann Hematol, 2010. 89(7): 691-699.
- Barbui T., et al: Sobrevivência e progressão da doença em trombocitmias essenciais são significativamente influenciadas pelo diagnóstico morfológico preciso: um estudo internacional. J Clin Oncol, 2011. 29(23): 3179-3184.
- Tefferi A, et al: Survival and prognosis among 1545 patients with contemporary polycythemia vera: an international study. Leucemia, 2013. 27(9): 1874-1881.
- Koschmieder S, et al.: Neoplasias mieloproliferativas e inflamação: quer para visar o clone maligno ou o processo inflamatório, quer ambos. Leucemia, 2016. 30(5): 1018-1024.
- Kroll MH, Michaelis LC, Verstovsek S: Mecanismos de trombogénese em policitemia vera. Blood Rev, 2015. 29(4): 215-221.
- Finazzi G, Barbui T: Evidência e perícia na gestão de policitemia vera e trombocitmia essencial. Leucemia, 2008. 22(8): 1494-1502.
- Mesa RA, et al: O fardo da fadiga e da qualidade de vida nas doenças mieloproliferativas (MPD): um inquérito internacional baseado na Internet de 1179 doentes com MPD. Cancro, 2007. 109(1): 68-76.
- Saini KS, Patnaik MM, Tefferi A: Policitemia vera-associated pruritus e a sua gestão. Eur J Clin Invest, 2010. 40(9): 828-834.
- Michiels JJ: Eritromelalgia e complicações vasculares em policitemia vera. Semin Thromb Hemost, 1997. 23(5): 441-454.
- De Stefano V, et al.: Splanchnic vein thrombosis and myeloproliferative neoplasms: molecular-driven diagnostic and long-term treatment. Thromb Haemost, 2016. 115(2): 240-249.
- Dentali F, et al: Trombose venosa cerebral e neoplasias mieloproliferativas: resultados de duas grandes bases de dados. Thromb Res, 2014. 134(1): 41-43.
- Elliott MA, Tefferi A: Trombose e hemorragia na policitemia vera e trombocitémia essencial. Br J Haematol, 2005. 128(3): 275-290.
- Michiels JJ, et al.: O paradoxo da activação plaquetária e da função deficiente: interacções plaquetário-von Willebrand factor, e a etiologia das manifestações trombóticas e hemorrágicas em trombocitmias essenciais e policitemia vera. Semin Thromb Hemost, 2006. 32(6): 589-604.
- Pardanani A, et al: Prevalência e correlatos clinicopatológicos de JAK2 exon 12 mutações em JAK2V617F-negativo policitemia vera. Leucemia, 2007. 21(9): 1960-1963.
- Vannucchi AM, et al.: correlatos clínicos da presença de JAK2V617F ou carga alélica nas neoplasias mieloproliferativas: uma reavaliação crítica. Leucemia, 2008. 22(7): 1299-1307.
- Passamonti F, et al.: Um estudo prospectivo de 338 pacientes com policitemia vera: o impacto da carga de alelos JAK2 (V617F) e da leucocitose na transformação de doenças fibróticas ou leucémicas e complicações vasculares. Leucemia, 2010. 24(9): 1574-1579.
- Ward AC, Touw I, Yoshimura A: O caminho Jak-Stat em hematopoiese normal e perturbada. Sangue, 2000. 95(1): 19-29.
- James C, et al: Uma mutação clonal única JAK2 que leva à sinalização constitutiva causa policitemia vera. Natureza, 2005. 434(7037): 1144-1148.
- Baxter EJ, et al: Mutação adquirida da tirosina kinase JAK2 em doenças mieloproliferativas humanas. Lancet, 2005. 365(9464): 1054-1061.
- Gangat N, et al.: Cytogenetic studies at diagnosis in polycythemia vera: clinical and JAK2V617F allele burden correlates. Eur J Haematol, 2008. 80(3): 197-200.
- Tefferi A, et al: Sequenciação profunda orientada em policitemia vera e trombocitémia essencial. Blood Adv, 2016. 1(1): 21-30.
- Scherber R, et al: The Myeloproliferative Neoplasm Symptom Assessment Form (MPN-SAF): ensaio internacional de validação prospectiva e fiabilidade em 402 doentes. Sangue, 2011. 118(2): 401-408.
- Grinfeld J, et al: Classificação e Prognóstico Personalizado em Neoplasias Mieloproliferativas. N Engl J Med, 2018. 379(15): 1416-1430.
- Marchioli R, et al: Eventos cardiovasculares e intensidade do tratamento em policitemia vera. N Engl J Med, 2013. 368(1): 22-33.
- Landolfi R, et al: Eficácia e segurança da aspirina de baixa dose em policitemia vera. N Engl J Med, 2004. 350(2): 114-124.
- Alvarez-Larran A, et al.: Observação versus terapia antiplaquetária como profilaxia primária para trombose em trombocitmias essenciais de baixo risco. Sangue, 2010. 116(8): 1205-1210; questionário 1387.
- Grunwald MR, et al: Clinical and Disease Characteristics From REVEAL at Time of Enrolment (Baseline): Estudo observacional prospectivo de pacientes com Policitemia Vera nos Estados Unidos. Clin Lymphoma Myeloma Leuk, 2018. 18(12): 788-795 e2.
- Michiels JJ, et al: eritromelárico, cerebral, ocular e coronário mediado por placas.
InFo ONCOLOGY & HEMATOLOGY 2019; 7(2-3): 12-15.









