As dermatoses auto-imunes bolhosas são um grupo clinicamente e imunopatologicamente heterogéneo de doenças auto-imunes raras. Em doentes idosos com prurido e bolhas salientes, o pemfigoide bolhoso está no topo da lista de diagnósticos diferenciais. Em doentes com erosões na área da mucosa oral (“estomatite aphtosa resistente à terapia”), possivelmente em combinação com bolhas/erosões na pele queratinizada, a presença de uma dermatose bolhosa auto-imune deve ser sempre considerada como um diagnóstico diferencial. A história da epistaxe, disfagia e dispneia é essencial neste caso, assim como uma inspecção detalhada da mucosa conjuntival e genital. O tratamento de dermatoses bulosas auto-imunes deve ser efectuado em centros especializados. Com novas opções terapêuticas tais como rituximab e imunoadsorção, mesmo as formas graves da doença podem ser bem tratadas na maioria dos casos. A determinação do antigénio alvo não só é importante para um diagnóstico exacto, como também pode indicar a presença de um processo paraneoplásico.
As dermatoses auto-imunes bolhosas são um grupo de doenças auto-imunológicas que têm em comum a presença de auto-anticorpos contra moléculas estruturais e de aderência da pele ou mucosa. Como resultado da desregulação tanto da imunidade humoral adaptativa como da imunidade celular, desenvolvem-se fendas que estão clinicamente associadas a bolhas e erosões na pele e/ou membranas mucosas.
Em relação à localização da formação da fenda, é possível dividir as dermatoses auto-imunes bolhosas em dois grupos principais para orientação:
1. Dermatoses bolhosas auto-imunes com perda de aderência intra-epitelial
– Doenças de pênfigo
2. dermatoses auto-imunes bolhosas com perda subepitelial de aderência
– Doenças pemfigoideas
– Dermatose linear de IgA
– Epidermólise bullosa acquisita
– Dermatite herpetiforme Duhring
Doenças de pênfigo
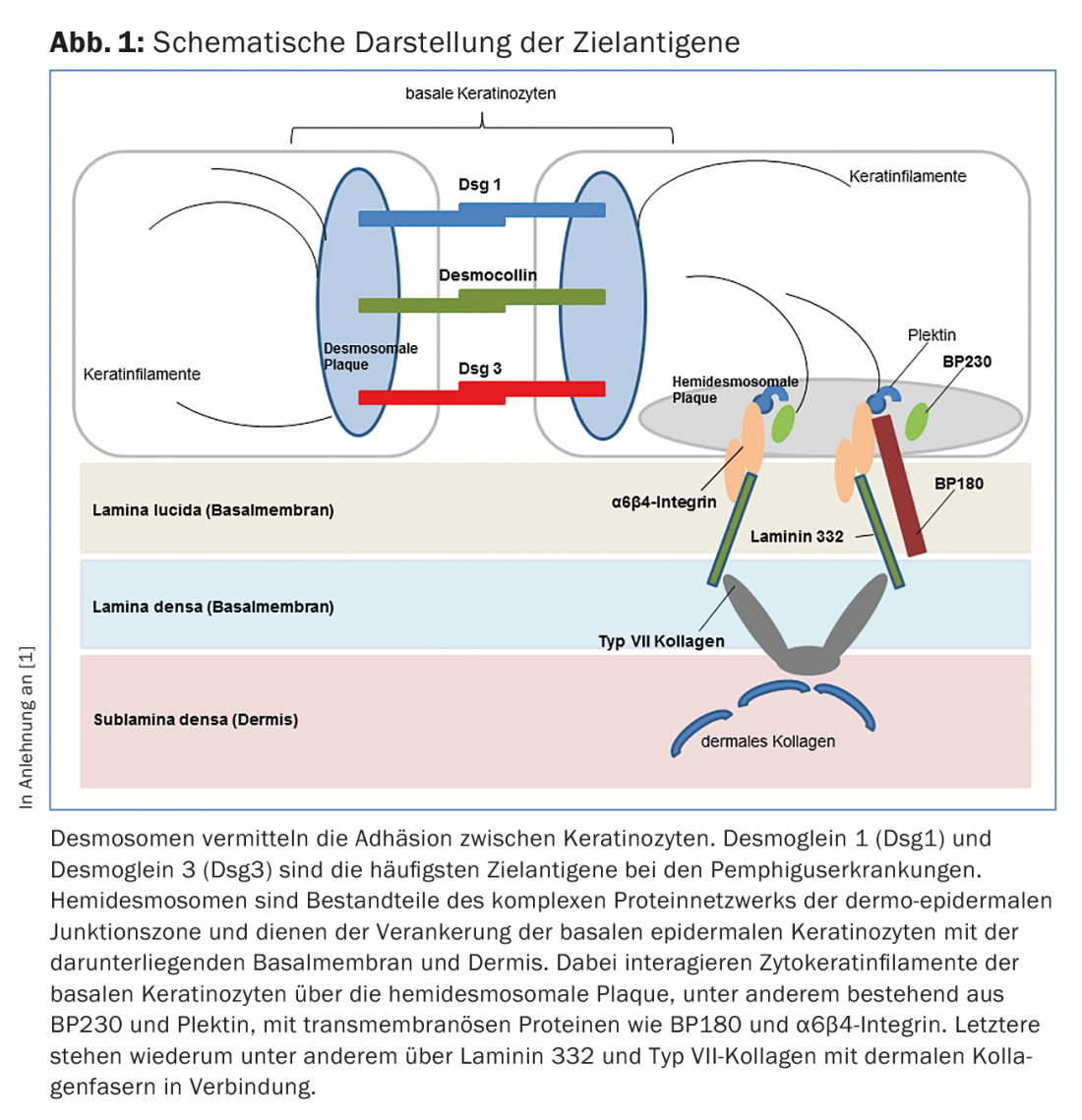
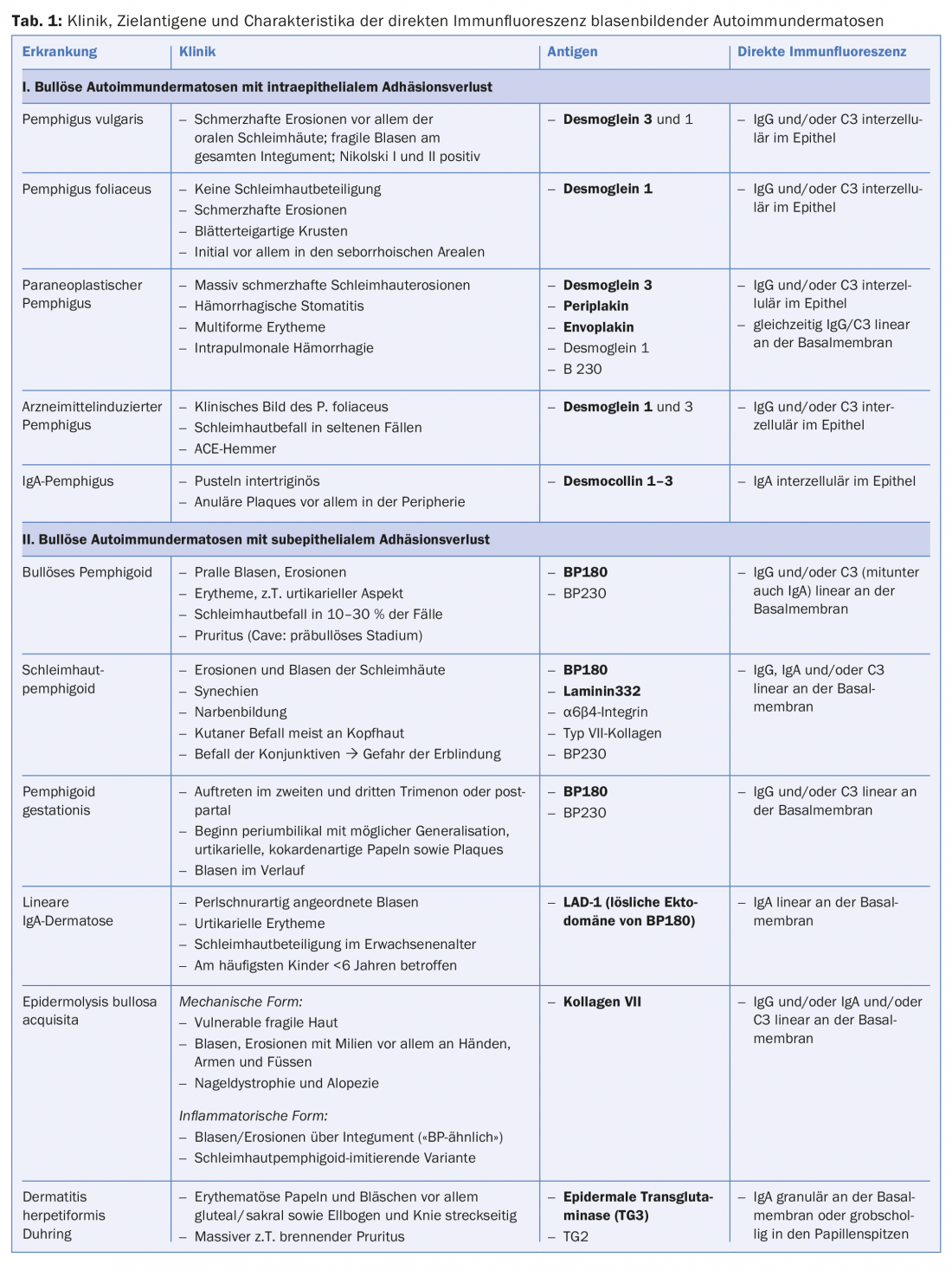
Nas doenças do grupo do pênfigo, há uma perda de contacto intra-epitelial de células devido a autoanticorpos contra proteínas dos desmosomas (Fig. 1), resultando numa formação de fenda intra-epidérmica localizada superficialmente. Clinicamente e imunopatologicamente, podem ser distinguidas diferentes formas de pênfigo.
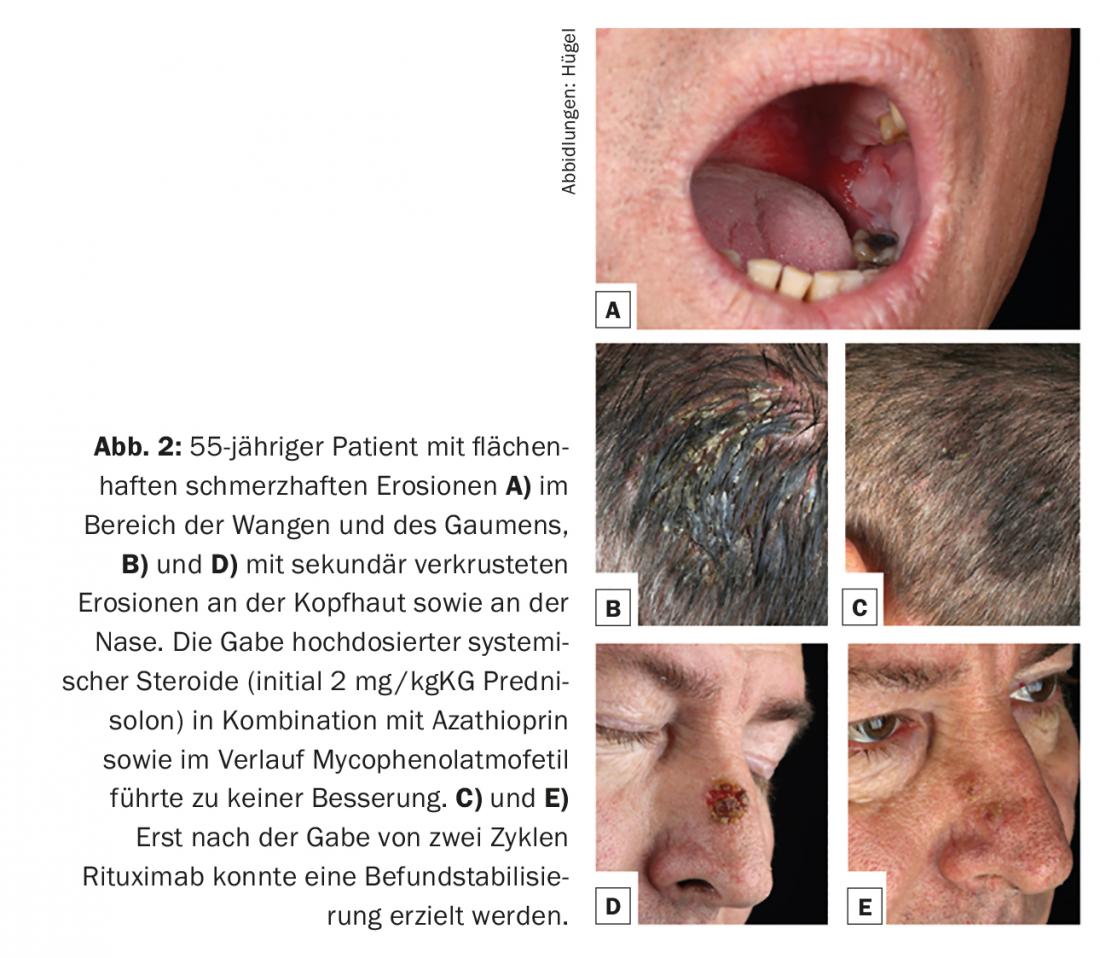
Pemphigus vulgaris representa aproximadamente 80% dos casos de pênfigo, manifestando-se preferencialmente entre a quarta e sexta décadas de vida, com uma incidência de 0,1-0,5/100,000/ano. Aetiopatogenicamente, autoanticorpos contra desmoglein 3 são sempre encontrados. No início da doença, há normalmente erosões da mucosa enoral muito dolorosas (Fig. 2A) e não é raro para a primeira apresentação a um especialista em doenças dos ouvidos, nariz e garganta. Opcionalmente, também se encontram anticorpos contra a desmoglein 1 em pemphigus vulgaris, com infestação consecutiva da pele queratinizada. (Fig. 2B e D). O aspecto clínico da pele é caracterizado por bolhas extremamente vulneráveis com um telhado de bolhas finas, que se rompem rapidamente e, por isso, normalmente já não se apresentam como bolhas, mas como (secundariamente incrustadas) erosões. O fenómeno Nikolski I (empurrabilidade das camadas epidérmicas superiores por tracção tangencial na pele intacta) pode ser induzido no exame clínico.
Em contraste, na segunda doença mais comum do pênfigo – pemphigus foliaceus – os auto-anticorpos só são encontrados contra a desmoglein 1, mas não contra a desmoglein 3. De acordo com o padrão de expressão das desmogleins, esta doença afecta, portanto, exclusivamente as áreas de pele córneas e não as membranas mucosas. Também aqui, quase nunca se encontram bolhas intactas, mas erosões extensivas com por vezes descamação em forma de folha. Os sintomas cutâneos começam frequentemente na cabeça peluda, rosto ou zona das ranhuras de suor frontal e posterior e depois espalham-se para a periferia.
O pênfigo paraneoplástico, que ocorre muito raramente, está associado em particular a malignidades hematológicas (especialmente linfomas não-Hodgkin de células B) e tem autoanticorpos contra alvos desmosomais e não-desmosomais. Clinicamente, caracteriza-se por extensas erosões dolorosas e úlceras das mucosas (especialmente boca, lábios, esófago), envolvimento conjuntival e lesões polimórficas da pele.
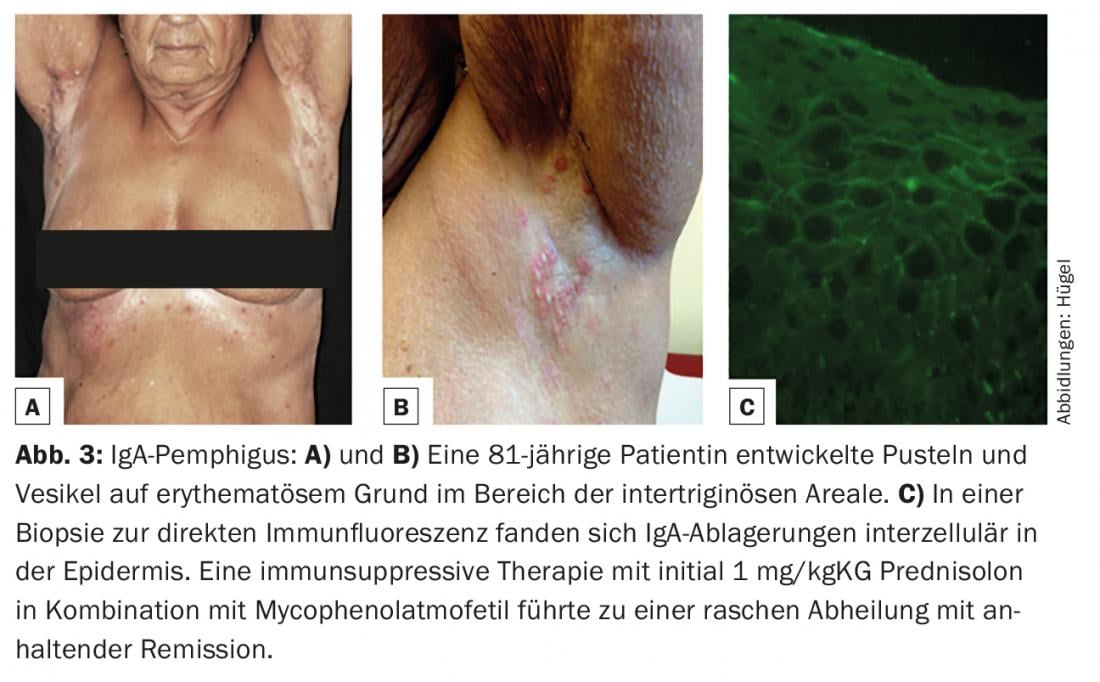
O pênfigo IgA (caracterizado por depósitos intraepidérmicos de IgA na biópsia da pele para imunofluorescência directa, Fig. 3C) é a mais rara das variantes do pênfigo. Clinicamente, pústulas e vesículas sobre uma base eritematosa encontram-se principalmente nas áreas intertriginosas (Fig. 3A e B).
Todos os doentes diagnosticados com pênfigo devem ter um historial de medicação cuidadoso, como medicamentos como a penicilamina (que, no entanto, é muito raramente utilizada no tratamento de doenças reumáticas actualmente), mas também os inibidores da ECA, como o captopril, enalapril ou lisinopril podem induzir a doença do pênfigo.
Dermatoses auto-imunes com perda subepitelial de aderência
Nas doenças pemfigoideas, as bolhas na pele aparecem muito mais estáveis e carnudas do que nas doenças pemphigus devido à formação de fendas subepidérmicas mais profundas. Os pacientes sofrem frequentemente de comichão grave. O quadro clínico é heterogéneo.
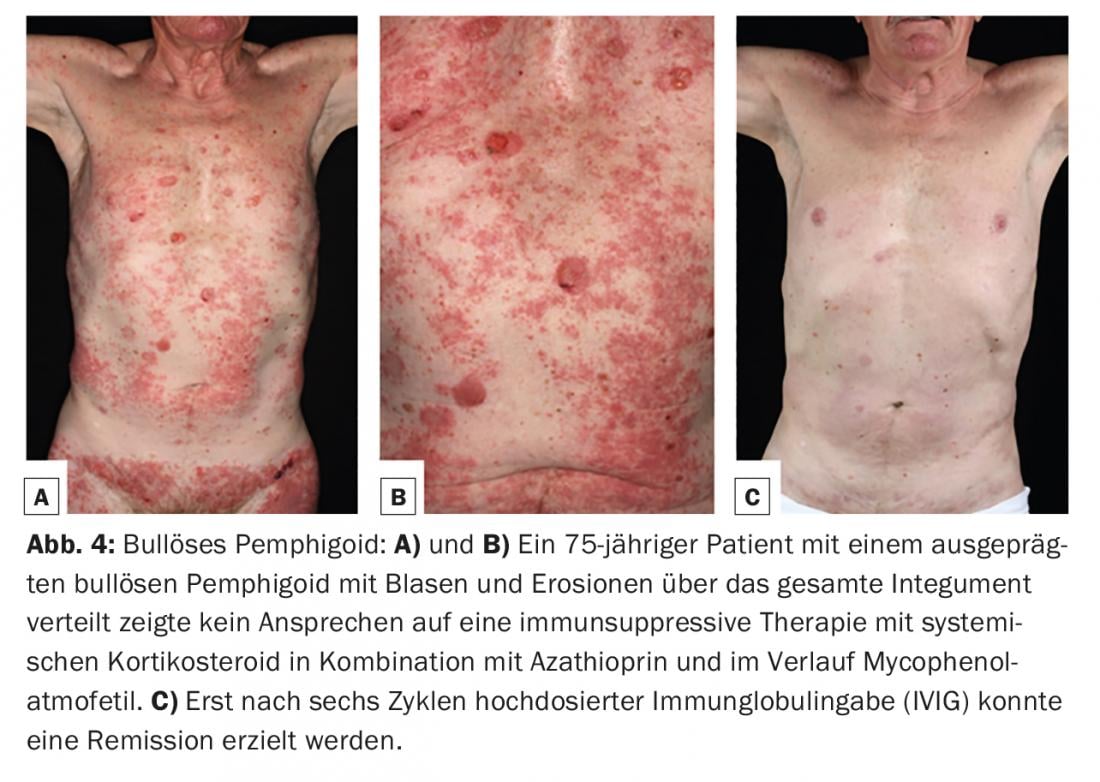
O pemfigoide bolhoso (PB) é a dermatose auto-imune bolhosa mais comum, com uma incidência de 12-21 casos por 1.000.000 de população/ano. A idade principal de início é entre os 60 e 90 anos de idade, e como resultado do aumento geral da esperança de vida, a incidência tem aumentado acentuadamente nos últimos anos. Uma característica do pemfigoide bolhoso é a presença de auto-anticorpos contra duas proteínas estruturais hemidematosas da zona da membrana basal: BP180 e/ou (mais raramente) BP230. (Fig.1). Clinicamente, o pemfigoide bolhoso apresenta-se com bolhas salientes cheias de líquido seroso, eritema disseminado e lesões urticárias seguidas de erosões e crostas. (Fig. 4A e B). As membranas mucosas são afectadas por 10-30%. Numa fase premonitória, a doença pode progredir durante meses a anos, sem borbulhar. Em doentes idosos com prurido grave e lesões cutâneas polimórficas (eczema foci, placas urticárias, escoriações como consequência do prurido), uma fase pré-bolhosa da PA deve, portanto, ser sempre considerada como um diagnóstico diferencial.
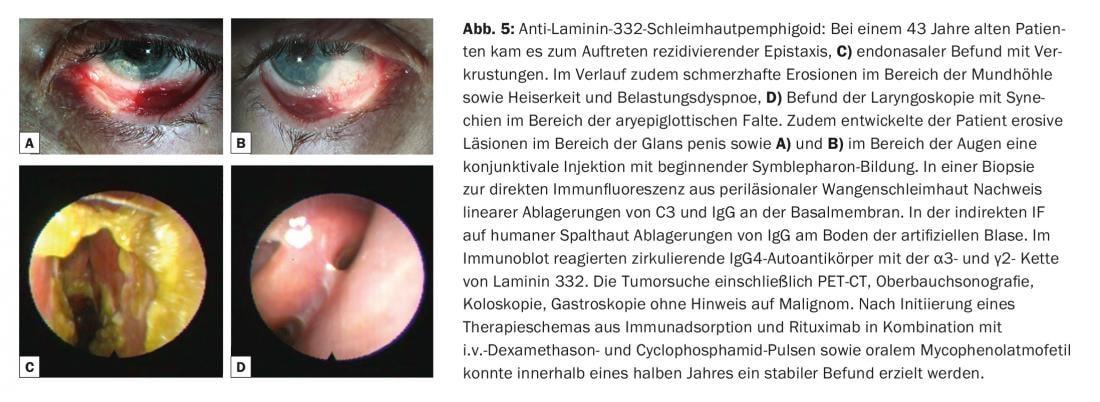
O termo pemfigoide da mucosa cobre um grupo heterogéneo de doenças raras do espectro das dermatoses auto-imunes bolhosas, que estão associadas a bolhas subepiteliais ou subepidérmicas e nas quais as alterações inflamatórias crónicas ocorrem predominantemente na área das mucosas. A formação de bolhas é causada por autoanticorpos contra moléculas de adesão da zona de junção dermo-epidérmica, e foram identificados pelo menos dez antigénios alvo diferentes. Na maioria das vezes, são encontrados auto-anticorpos IgG e/ou IgA circulantes contra a BP180. Menos frequentemente, autoanticorpos contra BP230, laminina-332, α6β4-integrina ou colagénio VII são detectados (Fig. 1). A identificação do antigénio alvo é de grande importância, uma vez que em 30% dos casos foi descrita uma associação com malignidades para um subtipo (anti-laminina-332 pemfigoide) e deve ser efectuada uma exclusão tumoral correspondente. Clinicamente, todas as membranas mucosas com epitélio escamoso estratificado podem ser afectadas no pemfigoide mucoso. O envolvimento mais frequente é o da mucosa oral, inicialmente com o aparecimento da gengivite desquamativa. O envolvimento ocular começa geralmente unilateralmente com o quadro clínico de conjuntivite, no decurso do qual há um encurtamento das dobras conjuntivais (fornices), simbolfáron, tricíase, sinéquias e atrofia da córnea com um eventual risco de cegueira. (Fig. 5A e B). A crosta endonasal, epistaxe, disfagia, rouquidão e estridor são encontrados com envolvimento nasofaríngeo ou envolvimento laríngeo. (Fig. 5C e D). Nos homens, as infestações genitais podem causar aderências entre o pénis da glande e o prepúcio, e nas mulheres, obstrução da vagina introitus. O envolvimento da pele – clinicamente semelhante ao pemfigoide bolhoso – é encontrado em cerca de 20% de todos os casos.
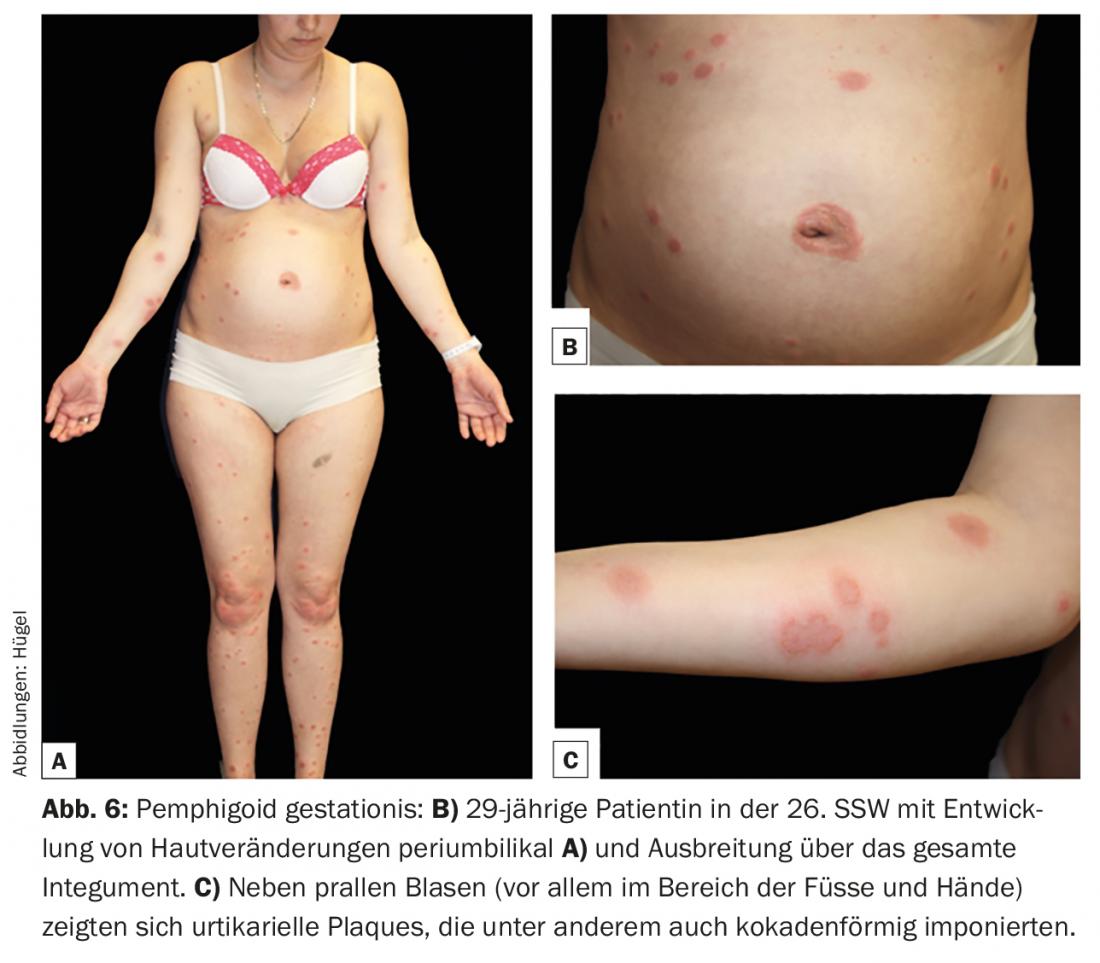
A gestação homofigoide (PG) é uma rara dermatose de gravidez que pode ocorrer no segundo e terceiro trimestres ou imediatamente pós-parto e é patofisiologicamente muito próxima do pemfigoide bolhoso. Tal como este, BP180 e – muito mais raramente – BP230 são os antígenos alvo cruciais. A causa para o desenvolvimento de auto-anticorpos ainda não foi esclarecida. O sistema HLA parece desempenhar um papel importante na predisposição genética. Discute-se que a apresentação de auto-antigénios placentários juntamente com moléculas paternais HLA classe II leva a uma indução de auto-anticorpos. Clinicamente, o desenvolvimento de eflorescências específicas é muitas vezes precedido por uma fase prodrómica com prurido intenso. As lesões cutâneas começam normalmente periumbilicamente (Fig. 6B) e podem depois espalhar-se por todo o tegumento. As lesões mais comuns são pápulas e/ou placas urticárias, aparecendo ocasionalmente em forma de coquetes. Apenas no decurso da doença se desenvolvem vesículas agrupadas ou bolhas salientes na maioria dos doentes (Fig. 6).
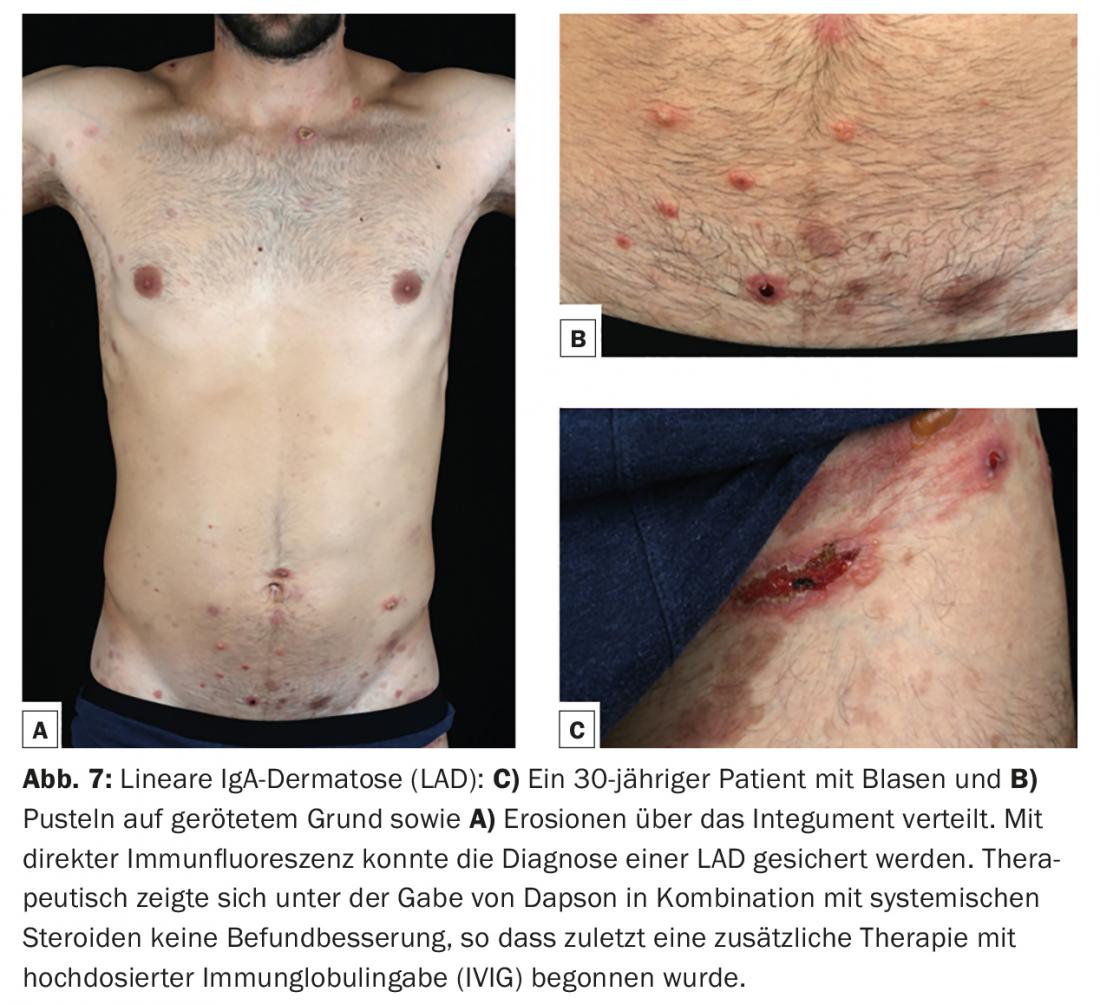
A dermatose linear de IgA (LAD) é a dermatose bula auto-imune mais comum da infância e depois começa normalmente antes dos seis anos de idade. Na idade adulta, a manifestação inicial é possível em qualquer idade, mas mais frequentemente após os 60 anos de idade. Tal como o pemphigoid de membrana mucosa, é considerado um quadro clínico heterogéneo. Os depósitos lineares do tipo IgA ao longo da zona de junção dermo-epidérmica na biópsia da pele para imunofluorescência directa são característicos. Os auto-anticorpos IgA são mais frequentemente dirigidos contra uma proteína de 97-kDa (LABD-97) e uma proteína de 120-kDa (LAD-1), que são produzidas por clivagem proteolítica da parte extracelular da BP180. Clinicamente, existem frequentemente bolhas anulares ou policíclicas na pele sã ou eritematosa com envolvimento preferencial do rosto (especialmente perioral e das orelhas), região anogenital e, menos frequentemente, tronco, mãos e pés. (Fig.7). A cicatrização das membranas mucosas, especialmente da conjuntiva, pode ocorrer, como no pemfigoide da mucosa, e pode mesmo levar à cegueira.
O quadro clínico da epidermólise bullosa acquisita (EBA), em que os anticorpos da classe IgG (e IgA) contra o colagénio VII estão etiopatogenicamente presentes (Fig. 1) , varia muito. Na variante mecanobolhosa, a pele é altamente vulnerável e encontram-se bolhas e erosões que cicatrizam cicatrizes ou com milia, especialmente em áreas mecanicamente estressadas tais como a parte de trás da mão, cotovelo, joelho, região sacral e dedos dos pés. Nas variantes inflamatórias, esta doença mostra um quadro clínico que pode ser difícil de distinguir do pemfigoide bolhoso ou do pemfigoide de membrana mucosa.
A dermatite herpetiforme Duhring é um subtipo cutâneo raro das doenças sensíveis ao glúten. Há alguns anos atrás, a transglutaminase epidérmica (transglutaminase 3) e transglutaminase de tecidos (transglutaminase 2) podem ser identificados. Os doentes com esta doença desenvolvem pápulas e vesículas principalmente nos lados extensores das extremidades, na cabeça cabeluda, glútea e sacralmente. Esta é uma condição extremamente irritante. Por conseguinte, a doença de Duhring deve ser considerada em todos os doentes com escoriações de arranhões em primeiro plano na área dos locais de predilecção acima mencionados.
Diagnóstico de dermatoses auto-imunes bolhosas
As biópsias de pele para exame histológico convencional podem ser utilizadas para mostrar a localização da fenda.
No entanto, o padrão de ouro de diagnóstico é a detecção de auto-anticorpos ligados aos tecidos (IgG, IgA e/ou factor complementar C3) em biópsias de pele ou mucosa com imunofluorescência directa. A localização dos anticorpos intercelulares na epiderme ou ao longo da zona de junção dermo-epidérmica permite uma diferenciação imediata entre a presença de uma doença pênfigo ou de uma doença do espectro das dermatoses auto-imunes subepiteliais bolhosas. Se predominarem precipitados do isótipo IgA, um pênfigo IgA (fig. 3C), uma dermatose linear IgA ou uma doença de Duhring podem ser diagnosticados, dependendo do padrão (tab. 1).
Para além das biópsias de pele (muco) e para a caracterização exacta dos diferentes subtipos, são necessários exames serológicos. A imunofluorescência indirecta no esófago dos macacos e na pele salina humana foi estabelecida como um teste de rastreio para este fim. Os antigénios alvo são então identificados com a ajuda de vários exames ELISA e, se necessário, exames de immunoblot específicos.
As concentrações de autoanticorpos no soro de doentes pemphigus e pemphigoidianos correlacionam-se geralmente bem com a actividade da doença e são, portanto, também adequadas para monitorizar a actividade da doença e avaliar a necessidade de mais terapia.
Terapia das dermatoses auto-imunes bolhosas
Em geral, a terapia das dermatoses auto-imunes com bolhas depende da gravidade, por um lado, e do subtipo diagnosticado, por outro. No caso, por exemplo, de um pemfigoide bolhoso localizado suave, o tratamento local apenas com esteróides de classe IV altamente potentes (pomada de propionato de clobetasol 0,05%) pode ser suficiente. Na maioria dos casos, contudo, a administração sistémica de corticosteróides em combinação com outros imunossupressores (por exemplo, azatioprina, micofenolato mofetil, ciclosporina, metotrexato, ciclofosfamida) é indicada. Dapsone é o agente de primeira linha para a dermatose linear IgA, dermatite herpetiforme Duhring e pemfigóide puro de membrana mucosa oral sem complicações. Em casos graves ou refractários, podem ser utilizadas imunoglobulinas intravenosas (IVIG), imunoadsorção e/ou rituximab. Além disso, vários relatórios de casos foram publicados nos últimos anos, demonstrando a eficácia da terapia com o anticorpo anti-IgE omalizumab em pemfigoide bolhoso. No entanto, isto requer estudos mais padronizados relativamente à dosagem, bem como à duração da utilização.
O tratamento de pacientes com dermatoses auto-imunes em bolhas deve ter lugar em centros especializados e requer – especialmente na presença de um subtipo com envolvimento da mucosa – uma abordagem interdisciplinar com envolvimento de oftalmologistas, otorrinolaringologistas, dentistas, ginecologistas, gastroenterologistas e o especialista em dermatologia como coordenador.
Literatura:
- Schmidt E, Zillikens E: Diagnóstico e terapia de dermatoses auto-imunes bolhosas. Dtsch Arztebl International 2011; 108(23): 399-405.
Leitura adicional:
- Hertl M (ed.): Doenças auto-imunes da pele. Patogénese, Diagnóstico, Gestão.3ª edição. Viena – Nova Iorque: Springer-Verlag 2011.
- Marazza G, et al: Incidência de pemfigoide bolhoso e pênfigo na Suíça: um estudo prospectivo de 2 anos. Br J Dermatol 2009; 161(4): 861-868.
- Kneisel A, Hertl M: Doenças auto-imunes de pele bolhosa. Parte 1: Manifestações clínicas. J Dtsch Dermatol Ges 2011; 9(10): 844-856.
- Kneisel A, Hertl M: Doenças auto-imunes de pele bolhosa. Parte 2: diagnóstico e terapia. J Dtsch Dermatol Ges 2011; 9(11): 927-947.
- Ajoelhar A, Hertl M: Penfigóide Bolhoso: diagnóstico e terapia. Wien Med Wochenschr 2014; 164(17-18): 363-371.
- Schmidt E, Zillikens D: Doenças pemfigoideas. Lancet 2013; 381(9863): 320-332.
- Hertl M, et al.: Recomendações para o uso de rituximab (anticorpo anti-CD20) no tratamento de doenças de pele bolhosas auto-imunes. J Dtsch Dermatol Ges 2008; 6(5): 366-373.
PRÁTICA DA DERMATOLOGIA 2017; 27(1): 18-25