A Queratose Folicular Espinulosa Decalvans, também conhecida como síndrome de Siemens, é uma genodermatose rara que afecta predominantemente os rapazes. Os sintomas clássicos são hiperqueratose folicular difusa em conjunto com alopecia cicatricial progressiva. Nalguns doentes, podem também ocorrer outros sintomas. É necessária uma abordagem interdisciplinar para uma gestão holística.
A primeira descrição da Queratose Folicular Espinulosa Decalvante (KFSD) data de 1926 [1]. A maioria dos casos é recessiva ligada ao X, mas também foram descritos casos esporádicos e autossómicos dominantes de KFSD [2]. A hereditariedade ligada ao X envolve mutações no gene SAT1 ou no gene MBTPS2. Para confirmar o diagnóstico de KFSD, são frequentemente necessárias análises histopatológicas, para além de testes genéticos. A manifestação inicial ocorre tipicamente na infância sob a forma de pápulas queratóticas, que aparecem primeiro na face e mais tarde se espalham para o tronco e membros [2]. À medida que a doença progride, desenvolve-se alopecia cicatricial, sendo também afectadas as sobrancelhas e as pestanas. As zonas axiais e genitais também apresentam queda de cabelo [4]. Alguns doentes desenvolvem sintomas associados, tais como fotofobia e distrofia corneana palmo-plantar, tendo também sido registada acne keloidalis nuchae [5].
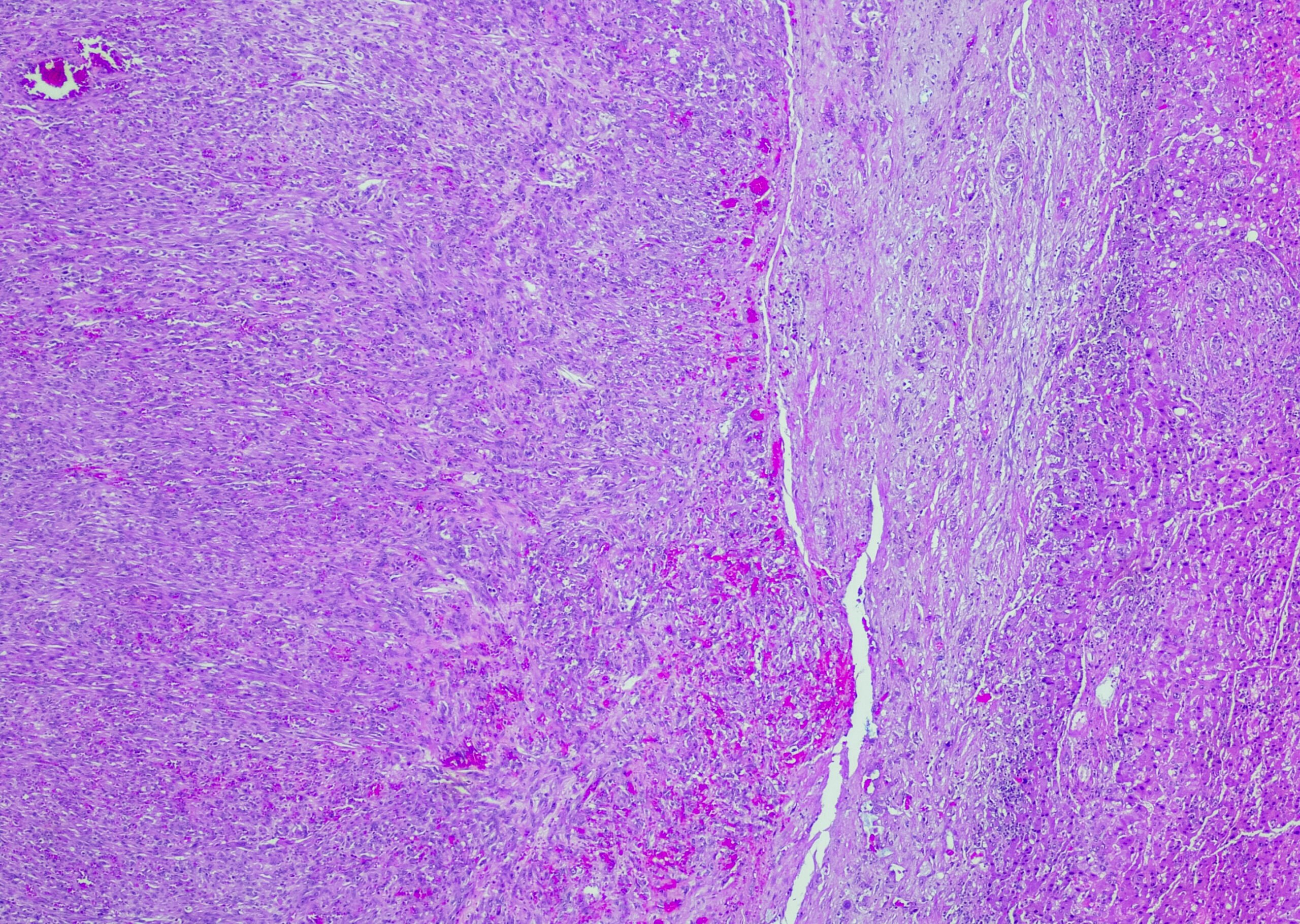
A histopatologia levou ao diagnóstico
A KFSD representa um desafio diagnóstico e terapêutico, como também foi demonstrado no presente caso de um paciente de 8 anos de idade [6].
Caraterísticas clínicas: O rapaz apresentava lesões inflamatórias no couro cabeludo e alopécia. Para além disso, o doente apresentava pápulas hiperqueratóticas difusas distribuídas por toda a superfície corporal, especialmente na área dos membros, bochechas e sobrancelhas. Não foi observada dismorfia facial nem fotofobia.
Diagnóstico: Os resultados dos testes genéticos moleculares foram inconclusivos, mas o diagnóstico clínico suspeito de KFSD foi confirmado por avaliação histopatológica de uma biopsia do couro cabeludo.
Tratamento: O doente foi tratado com isotretinoína administrada por via sistémica, clindamicina tópica e emolientes, o que levou a uma redução da inflamação perifolicular e a uma redução da queratose, estabilizando a queda de cabelo.
Comentário: Histopatologicamente, revelou-se útil para excluir diagnósticos diferenciais, o que apoiou a KFSD como o diagnóstico correto, sendo o diagnóstico baseado principalmente nas caraterísticas clínicas.
DD: Síndrome IFAP e GLPLS
Os diagnósticos diferenciais mais importantes incluem a síndrome de ictiose folicular alopécia fotofóbica (IFAP) e a síndrome de Graham-Little-Piccardi-Lasseur (GLPLS) [1]. A síndrome IFAP é uma doença alélica com semelhanças genotípicas e fenotípicas com a KFSD. Ambas são manifestações fenotípicas diferentes de uma mutação no gene MBTPS2 [7]. Clinicamente, ambas as doenças são caracterizadas por alopecia e hiperqueratose folicular, sendo que a síndrome IFAP, ao contrário da KFSD, é uma alopecia não cicatricial. Além disso, as manifestações oculares, como a fotofobia e a distrofia da córnea, são caraterísticas essenciais da síndrome IFAP [8]. A GLPLS é uma variante do líquen plano pilar e caracteriza-se por alopecia cicatricial da cabeça e alopecia não cicatricial das axilas e áreas genitais, acompanhada por pápulas foliculares queratóticas distribuídas pelo corpo. A diferenciação da KFSD baseia-se principalmente na histopatologia, especialmente porque a GLPLS tem caraterísticas clássicas de líquen plano [3].
O tratamento do KFSD revela-se frequentemente difícil. Os queratolíticos e os emolientes são utilizados principalmente para o tratamento tópico. Os retinóides sistémicos, como a isotretinoína, demonstraram ser úteis nas fases iniciais do tratamento, reduzindo a hiperqueratose folicular e a inflamação [4]. A dapsona mostra alguma eficácia devido à inibição da quimiotaxia dos leucócitos e à estabilização das enzimas lisossómicas [9]. Outras opções de tratamento incluem antibióticos, bem como esteróides intralesionais e tópicos [9].
Congresso: Congresso Anual da SGDV
Literatura:
- Malvankar DD, Sacchidanand S: Queratose Folicular Espinulosa Decalvans: Relato de Três Casos. Int J Trichology 2015; 7(3): 125-128.
- Bellet JS, et al: Queratose folicular espinulosa decalvante numa família. JAAD 2008; 58: 499-502.
- Brar B, Khanna E, Mahajan BB: Síndrome de Graham little piccardi lasseur: Um relato de caso raro com líquen plano hipertrófico concomitante. Int J Trichology 2013; 5: 199-200.
- Sequeira FF, Jayaseelan E: Queratose folicular espinulosa decalvante numa mulher. Indian J Dermatol Venereol Leprol 2011; 77: 325-327.
- Janjua SA, et al: Queratose folicular espinulosa decalvante associada a acne keloidalis nuchae e foliculite pilosa em tufos. Am J Clin Dermatol 2008; 9: 137-140.
- Bianchetti L, et al: Relato de caso: Queratose Folicular Espinulosa Decalvans num rapaz de 8 anos de idade. Poster 111, Congresso Anual da SGDV, 18-20 de setembro de 2024.
- Aten E, et al: A queratose folicular espinulosa decalvante é causada por mutações no MBTPS2. Hum Mutat 2010; 31: 1125-1133.
- Alfadley A, et al: Dois irmãos com queratose folicular espinulosa decalvante. JAAD 2002;47: 275-278.
- Kunte C, Loeser C, Wolff H: Foliculite espinhosa decalvante: Terapia bem sucedida com dapsona. JAAD 1998; 39: 891-893.
DERMATOLOGIE PRAXIS 2024; 34(6): 29 (publicado em 13.12.24, antes da impressão)