A síndrome, descrita pela primeira vez por dois médicos, o Dr. John Crigler e o Dr. Victor Najjar, em 1952, caracteriza-se por uma perturbação congénita do metabolismo da bilirrubina. As opções de tratamento existentes têm como objetivo reduzir a quantidade de bilirrubina não conjugada no sangue. No entanto, isto requer, por vezes, procedimentos relativamente complexos. As alternativas incluem um transplante de fígado ou, eventualmente, um transplante de hepatócitos. Mas há uma luz no horizonte: uma terapia genética obteve resultados promissores num ensaio clínico.
A bilirrubina é um produto de degradação do pigmento vermelho do sangue, a hemoglobina, e forma-se quando os glóbulos vermelhos se decompõem. [1–3]Normalmente, a enzima UGT1A1 (UDP-glucuronosiltransferase 1 polipeptídeo A1) catalisa a formação do diglucuronídeo de bilirrubina solúvel em água no retículo endoplasmático liso do fígado, que é depois excretado para o intestino através dos canais biliares. No entanto, as pessoas afectadas pela síndrome de Crigler-Najjar não possuem esta enzima, o que faz com que a bilirrubina se acumule no corpo e, sem tratamento, leve a danos neurológicos significativos ou mesmo à morte [1]. Na síndrome de Crigler-Najjar tipo 1, a enzima UGT está completamente inativa e no tipo 2 está muito reduzida [4]. Ambas as formas são causadas por defeitos genéticos no gene UGT1A1 no cromossoma 2. Devido à natureza genética da doença, ambos os pais têm de ser portadores da mutação para que o seu filho seja afetado [2]. Estima-se que menos de 1 em 1 milhão de recém-nascidos em todo o mundo seja afetado pela síndrome de Crigler-Najjar.
Aspeto clínico
A síndrome de Crigler-Najjar tipo 1 manifesta-se normalmente logo após o nascimento como hiperbilirrubinemia excessiva, que, se não for tratada, conduz normalmente a kernicterus com danos neurológicos graves. Como resultado, os doentes afectados morrem frequentemente na primeira infância se não forem tratados. [11]Os recém-nascidos são particularmente susceptíveis a danos neurológicos causados pela hiperbilirrubinemia, uma vez que o fígado, ainda em desenvolvimento, é fortemente pressionado pela degradação da hemoglobina fetal nos primeiros dias de vida e a barreira hemato-encefálica ainda não está muito bem desenvolvida. Níveis séricos de bilirrubina ligeiramente elevados e iterícia não são invulgares nos recém-nascidos, mas os níveis de bilirrubina devem ser cuidadosamente monitorizados se houver qualquer sinal de aumento.
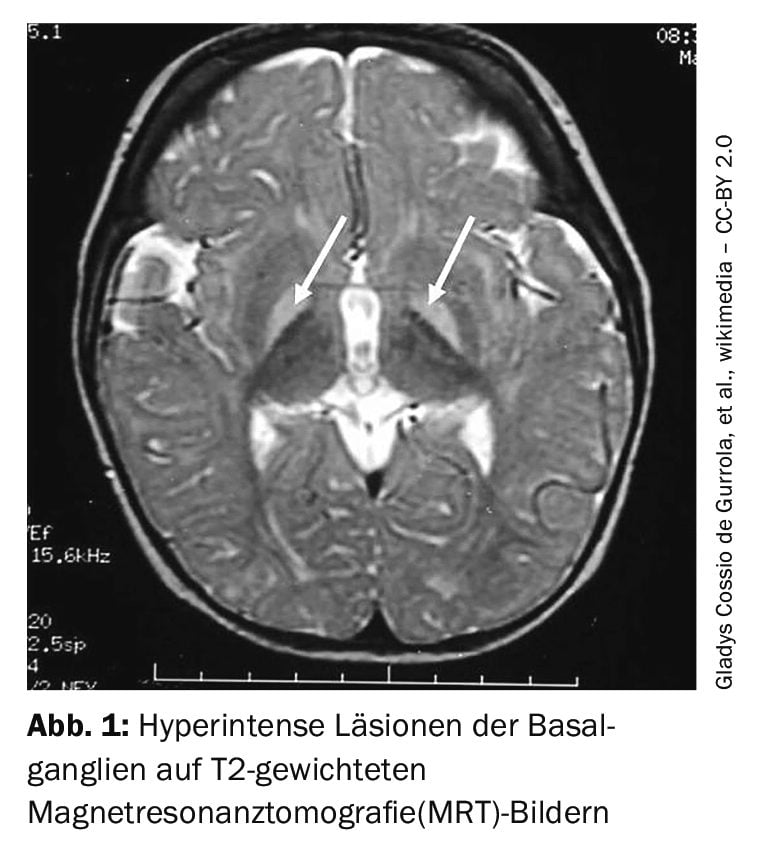
A síndrome de Crigler-Najjar tipo 2 é menos grave do que o tipo 1, onde o kernicterus é raro, mas os sintomas perturbadores com amarelecimento da pele e prurido extenso podem afetar gravemente a qualidade de vida [4]. Nalgumas pessoas, o diagnóstico não é feito até à idade adulta, como é o caso do relato de caso resumido na caixa [5]. O kernicterus é raro no tipo 2, mas pode ocorrer especialmente quando a pessoa afetada está doente, sem comer ou sob anestesia [2]. Se a iterícia grave poucos dias após o nascimento pode ser a síndrome de Crigler-Najjar, pode ser confirmado pela avaliação clínica, história familiar e testes genéticos e laboratoriais. Um achado clássico seria, por exemplo, um nível elevado de bilirrubina não conjugada no sangue ou uma falta de bilirrubina conjugada na bílis. Os testes genéticos para identificar mutações no gene UGT1A1 podem confirmar o diagnóstico [2].
Relato de caso: diagnóstico de SNC tipo 2 na idade adulta Um homem de 21 anos era afetado por episódios recorrentes de iterícia desde a infância. Nos últimos 6 meses, tinha sido particularmente afetado por iterícia persistente, acompanhada de vómitos ocasionais. |
| Historial: O seu nascimento decorreu sem complicações na altura, não houve iterícia neonatal nem necessidade de transfusões de sangue. Atingiu os marcos de desenvolvimento esperados para a sua idade e não houve motivos de preocupação. Aos 5 anos, os seus pais notaram pela primeira vez uma estranha descoloração amarelada dos olhos, que não era acompanhada de febre, comichão, dores abdominais ou fezes cor de barro, mas a urina era fortemente descolorida. O doente submeteu-se então a vários tratamentos alternativos e complementares. Infelizmente, nenhuma destas medidas conduziu a uma cura completa do seu estado. Segundo as recordações dos pais do doente, o nível mais elevado de bilirrubina sérica alguma vez medido foi de 12 mg/dL. |
| Investigações de diagnóstico actuais: O exame atual revelou iterícia sem organomegalia. Outras investigações clínicas também revelaram resultados normais e não ofereceram qualquer explicação imediata para a sua iterícia persistente. No entanto, as investigações de rotina revelaram hiperbilirrubinemia indireta com enzimas hepáticas normais. O exame ultrassonográfico do abdómen não apresentava alterações. Foi então efectuado um exame hemolítico completo, mas todos os testes foram negativos. Com base no início, na evolução e na presença de hiperbilirrubinemia não conjugada, foi feito um diagnóstico provisório de hiperbilirrubinemia não hemolítica não conjugada. Com a suspeita de uma síndrome de bilirrubinemia indireta congénita, foi iniciada uma análise para mutações UGT1A1, que foi positiva e indicou a presença de uma deficiência parcial da enzima. Este facto levou ao diagnóstico definitivo de síndrome de Crigler-Najjar tipo 2. |
| Terapêutica: Para controlar os sintomas do doente e melhorar a sua qualidade de vida, foi-lhe administrado fenobarbital oral numa dose de 5 mg/kg. De forma notável, observou-se uma redução significativa dos níveis de bilirrubina sérica do doente apenas duas semanas após o início do tratamento. Assim, a terapêutica revelou-se eficaz. |
| de acordo com [5] |
Opções terapêuticas atualmente disponíveis
O principal objetivo do tratamento de doentes com síndrome de Crigler-Najjar é reduzir a quantidade de bilirrubina não conjugada no sangue tão rápida e consistentemente quanto possível. Isto é recomendado para a síndrome de Crigler-Najjar tipo 1 (CNS I) e tipo 2 (SNC II) de formas diferentes [2].
O tratamento conservador do SNC I baseia-se em três pilares [4]:
- Fototerapia diária consistente com luz azul (torna a bilirrubina hidrossolúvel)
- Administração de tinprotoporfirina, um inibidor da hemoxigenase (reduz as elevações da bilirrubina)
- Administração de carbonato de cálcio e fosfato de cálcio (aumenta a secreção de bilirrubina não conjugada no intestino)
Esta terapia pode prolongar a esperança de vida e atrasar o aparecimento de complicações neurológicas. Uma outra opção terapêutica, o transplante de fígado, deve ser tentada o mais cedo possível. O transplante alogénico de hepatócitos encontra-se atualmente em fase experimental.
O SNC II é tratado com a administração de fenobarbital uma vez por dia [4]. Em alternativa, a rifampicina também é possível. Ao induzir a atividade enzimática, a concentração de bilirrubina no plasma pode ser reduzida para níveis seguros.
Estudo da terapia génica: oportunidades e riscos D’Antiga et al. [6,7]investigaram a segurança e a eficácia de uma infusão intravenosa única de um vetor AAV que codifica a UGT1A1 em 5 doentes com síndrome de Crigler-Najjar . |
| Em três dos doentes tratados com uma dose mais elevada, os níveis de bilirrubina desceram para menos de 30 µmol por litro (17,5 mg/dl), o que significa que a fototerapia podia ser descontinuada durante os 18 meses seguintes de seguimento [7]. No entanto, a normalização completa do nível de bilirrubina não foi alcançada em nenhum caso. |
| De acordo com Di Dato et al. [6,8]a duração da eficácia de uma única infusão do vetor AAV ainda não é clara. Além disso, foi referido que, em doentes com hemofilia, a terapêutica genética pode levar ao desenvolvimento de anticorpos AAV neutralizantes persistentes, de elevado título e com reação cruzada, o que poderia excluir a possibilidade de novas administrações do vetor [9]. [10]As infusões múltiplas de vectores AAV podem também representar um risco de genotoxicidade. |
A terapia genética como potencial método de tratamento alternativo
A utilização de vectores AAV (Vírus Adeno-Associado), por exemplo, foi aprovada pela Food and Drug Administration (FDA) dos EUA para a terapia de substituição genética em doentes com atrofia muscular espinal e cegueira congénita. [1,7]Os resultados iniciais de um ensaio clínico sobre a síndroma de Crigler-Najjar sugerem que a terapia genética baseada em AAV pode constituir um potencial tratamento alternativo para esta doença potencialmente fatal. O tratamento, que está atualmente a ser testado, foi desenvolvido por investigadores da Généthon**. Trata-se de fornecer às células do fígado uma cópia do gene UGT1A1, que codifica uma enzima que facilita a eliminação da bilirrubina. As primeiras observações do estudo CureCN (“Adeno-Associated Virus Vetor-Mediated Liver Gene Therapy for Crigler-Najjar Syndrome”) sugerem que a terapia genética pode ser uma potencial alternativa de tratamento. [Adeno-assoziiertes Virus] “Estamos muito entusiasmados com os resultados alcançados até agora neste estudo de terapia genética mediada por AAV para o tratamento da síndrome de Crigler-Najjar”, observou o Dr. D’Antiga [1]. “O tratamento demonstrou ser seguro em doses adequadas e capaz de afetar a doença de tal forma que o primeiro doente pôde interromper a sua fototerapia diária, eliminando o risco de danos neurológicos. O grau de melhoria da segunda doente sugere que também ela poderá em breve suspender a fototerapia” [1].
** A Genethon faz parte do Instituto de Bioterapias para as Doenças Raras (BIRD)
Um artigo publicado em 2024 por Di Dato et al. [6] também avalia a terapia génica como uma potencial alternativa de tratamento promissora, embora os autores refiram que ainda existem algumas questões por responder relativamente à eficácia e segurança desta abordagem de tratamento, que são objeto de investigações actuais (caixa).
Literatura:
- “Estudo clínico dá esperança a pessoas que sofrem de uma doença hepática genética rara”, https://cordis.europa.eu/article/id/430456-clinical-trial-gives-hope-to-sufferers-of-rare-genetic-liver-disease/de,(último acesso em 29/08/2024).
- “Crigler-Najjar syndrome”, https://liverfoundation.org,(último acesso em 29/08/2024).
- “UDP-glucuronyltransferase mutation (UGT1A1*28)”,www.labor-duesseldorf.de/examination/view/udp-glukuronyltransferase-mutation-ugt1a128,(último acesso em 29/08/2024).
- “Crigler-Najjar syndrome”, https://flexikon.doccheck.com,(último acesso em 29/08/2024).
- Rijal D, et al: Um caso raro de síndrome de Crigler-Najjar tipo 2: Um relato de caso e revisão da literatura. Clin Case Rep 2023; 13 de novembro; 11(11): e8176.
- Di Dato F, D’Uonno G, Iorio R: Síndrome de Crigler-Najjar: olhar para o futuro não nos faz esquecer o presente. Orphanet J Rare Dis. 2024 Mar 7; 19(1): 102.
- D’Antiga L, et al: Terapia genética em pacientes com a síndrome de Crigler-Najjar. NEJM 2023; 389(7): 620-631.
- Aronson SJ, Ronzitti G, Bosma PJ: O que se segue na terapia genética para a síndrome de Crigler-Najjar? Expert Opin Biol Ther 2023; 23(2): 119-121.
- George LA, et al: Acompanhamento a longo prazo da primeira entrega intravascular em humanos de AAV para transferência de genes: AAV2-hFIX16 para hemofilia B grave. Mol Ther 2020; 28 (9): 2073-2082.
- Sabatino DE, et al: Avaliação do estado da ciência da integração do vírus adeno-associado: uma perspetiva integrada. Mol Ther 2022; 30(8): 2646-2663.
- Wikipédia: Kernicterus, https://en.wikipedia.org,(último acesso em 29/08/2024).
GP PRACTICE 2024; 19(9): 44-45