Esta síndrome muito rara é causada por mutações no gene LRP2, que codifica a megalina, e é herdada de forma autossómica recessiva. Caracteriza-se por caraterísticas craniofaciais com traços faciais caraterísticos. Para além das complicações oculares e da perda auditiva neurossensorial, estão descritas outras anomalias clínicas. O diagnóstico suspeito pode ser confirmado através de testes genéticos. Dependendo da evolução individual, são necessárias várias medidas terapêuticas.
A síndrome de Donnai-Barrow (DBS) está associada a múltiplas malformações congénitas [1]. Os indivíduos afectados caracterizam-se por dismorfias faciais típicas, miopia e outros achados oculares, bem como perda de audição, agenesia do corpo caloso, proteinúria de baixo peso molecular e várias perturbações do desenvolvimento intelectual. Uma hérnia diafragmática congénita e/ou uma onfalocele são comuns. Existem poucos dados disponíveis sobre a prevalência e a incidência da DBS. Até à data, foram descritos menos de 50 indivíduos afectados de cerca de 20 famílias. A DBS ocorre em todos os grupos étnicos e não parece haver um gradiente de género. A DBS é causada por mutações no gene LRP2 (proteína 2 relacionada com o recetor de lipoproteínas de baixa densidade; 2q31.1). Este gene codifica a megalina, que é expressa em vários epitélios de reabsorção – particularmente no cérebro, nos rins e nos olhos. A megalina desempenha um papel importante em várias cascatas de sinalização e na endocitose de muitos ligandos.
Caraterísticas clínicas comuns
Quase todos os doentes apresentam os seguintes sintomas [1]:
- Agenesia/hipogénese do corpo caloso
- Fontanela anterior alargada
- perda auditiva neurossensorial acentuada (ou seja, o som chega ao ouvido interno mas não pode ser convertido em impulsos nervosos ou estes não são transmitidos ao cérebro)
- Hipertelorismo.
Os traços faciais caraterísticos são [1]:
- fissuras palpebrais inclinadas para baixo,
- Nariz curto com uma ponte nasal plana,
- Testa larga e alta
- “pico de viúva” na linha do cabelo da frente e, por vezes, proptose
Cerca de 40% dos doentes têm uma hérnia diafragmática congénita e/ou uma onfalocele. O desenvolvimento é frequentemente atrasado e a inteligência é reduzida em graus variáveis. A miopia elevada (>6 dptr) pode levar a descolamento/distrofia da retina e a um aumento da deficiência visual. Ocasionalmente, foram descritos coloboma da íris, glomerulosclerose segmentar focal e disfunção dos túbulos proximais (raramente levando a insuficiência renal).
Diagnóstico e DD O diagnóstico da síndrome de Donnai-Barrow (DBS) é feito através de uma combinação de caraterísticas clínicas e imagiológicas, juntamente com um padrão típico de proteinúria de baixo peso molecular, níveis urinários elevados de proteína de ligação ao retinol (RBP) e rácio RBP/creatinina. O diagnóstico é confirmado por uma análise do ADN. Devido à constelação caraterística de sintomas, o número de diagnósticos diferenciais da DBS é limitado. Alguns sintomas comuns incluem a tetrassomia 12p, bem como os síndromes de Fryns, Chudley-McCullough, acrocallosal e crânio-fronto-nasal. O fenótipo renal é parcialmente semelhante à doença de Dent e à síndrome de Lowe. O fenótipo ocular pode indicar o síndroma de Stickler. A deteção de hipertelorismo e hérnia diafragmática congénita ou onfalocele em imagiologia pré-natal deve sugerir DBS. O diagnóstico pré-natal em gravidezes de alto risco requer a identificação prévia das mutações causadoras da doença na família. |
| de acordo com [1] |
Tratamento terapêutico e prognóstico
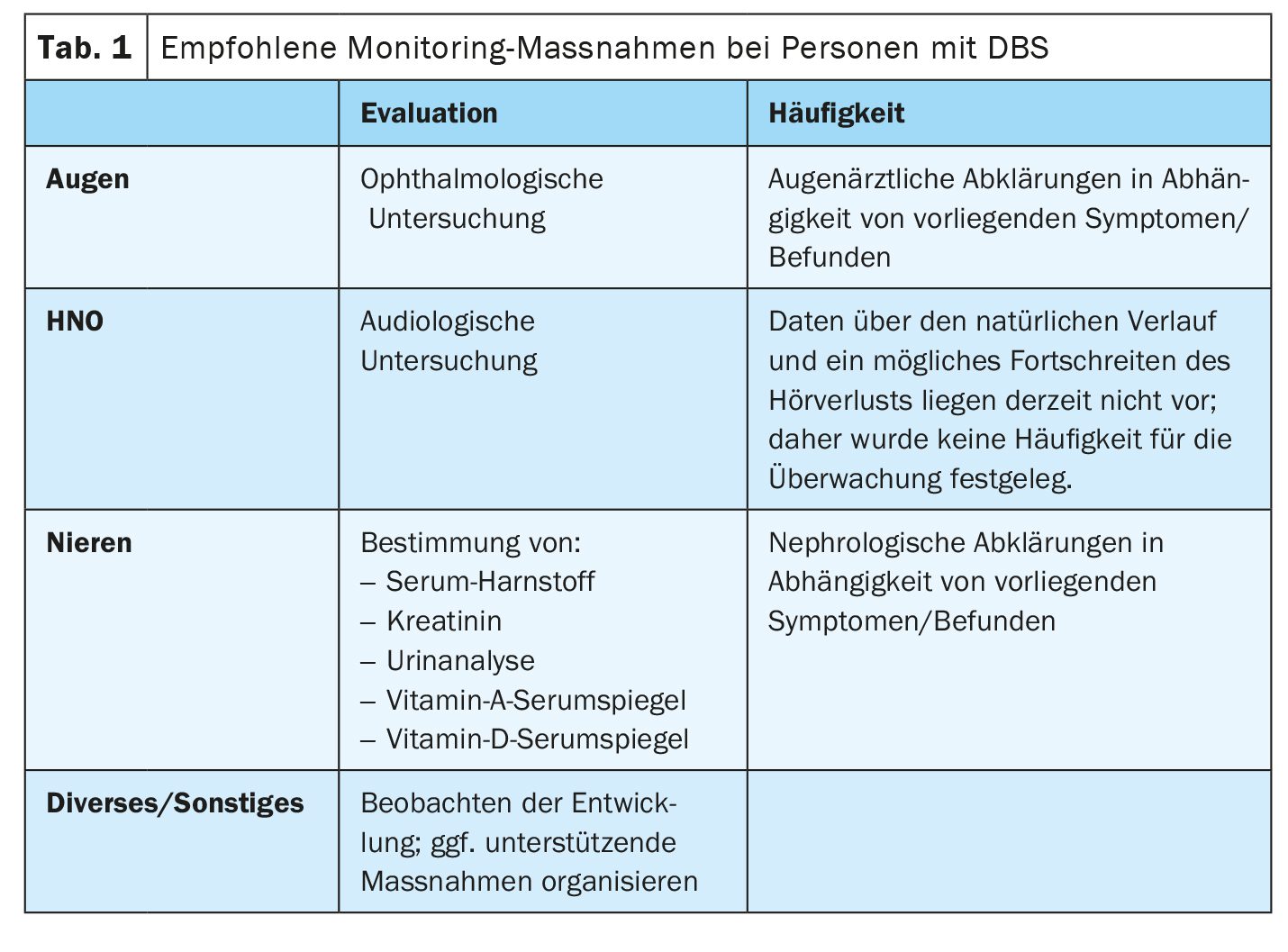
A DBS é herdada de uma forma autossómica recessiva. Deve ser oferecido aconselhamento genético aos pais das crianças afectadas e aos seus familiares [1]. Com exceção de um único caso publicado de dissomia uniparental, os pais dos doentes cujos casos foram documentados eram heterozigotos obrigatórios. São necessários controlos regulares da acuidade visual, da audição e da função renal. A correção dos óculos, o tratamento para evitar o descolamento da retina, os aparelhos auditivos e/ou os implantes cocleares fazem parte do plano de tratamento. A hérnia diafragmática congénita e/ou a onfalocele podem exigir intervenção cirúrgica. Devem ser propostas medidas de apoio específicas para as crianças afectadas. As pessoas afectadas podem alcançar uma visão e audição utilizáveis com correção. O estado de saúde geral dos doentes na infância e na adolescência é geralmente bom. A insuficiência renal terminal é uma complicação rara e potencialmente fatal. A apresentação pré ou perinatal com defeitos diafragmáticos e da parede abdominal requer intervenção cirúrgica e está associada a uma maior morbilidade e mortalidade.
Estudo de caso: progressão desde o nascimento até à idade da escola primária
Um rapaz atualmente com 9 anos de idade nasceu de pais caucasianos saudáveis, não aparentados, que tinham 34 anos (mãe) e 40 anos (pai) na altura [2]. Tinha uma irmã saudável e dois meios-irmãos saudáveis do lado da mãe. Durante a gravidez, um pequeno exomphalos foi detectado por ultrassom. O parto foi normal. Os exames clínicos e de imagem pós-natais revelaram hipertelorismo pronunciado, coloboma bilateral (formação de fenda na área dos olhos), ausência do corpo caloso, má rotação do intestino, hérnias inguinais bilaterais, mas nenhuma hérnia diafragmática congénita.
Onfalocele: A hérnia umbilical foi reduzida no primeiro dia, as suas hérnias foram reparadas com 1 ano de idade e a má rotação foi finalmente operada aos 18 meses de idade.
Agenesia do corpo caloso: Aos 4 meses de idade, o perímetro cefálico do doente era de 44,5 cm (percentil 95), a altura de 60,6 cm (percentil 90) e o peso de 5,78 kg (percentil 90). A ressonância magnética do cérebro confirmou a agenesia do corpo caloso e mostrou também uma encefalocele frontal com uma fossa anterior alargada e uma malformação de Chiari 1 com amígdalas cerebelares que se estendiam até C1.
Manifestações oculares: Para além dos colobomas da íris e coriorretinianos bilaterais, apresentava miopia elevada, catarata inferior direita e lenticonus posterior esquerdo, diagnosticados aos 3 meses de idade. A miopia estava associada a globos oculares aumentados (comprimento axial de 30 mm aos 7 anos de idade) e a estafilomas posteriores bilaterais, tendo sido submetido a retinopexia laser profiláctica de 360 graus para evitar o descolamento da retina. A sua prescrição de óculos era OD (Oculus Dexter) -15.00 D, OS (Oculus Sinister) -19.25/-2.00 eixo 92°, embora usasse normalmente lentes de contacto e conseguisse uma acuidade visual corrigida de OD 20/200, OS 20/100. Aos 7 anos de idade, o doente apresentava resultados de eletrodiagnóstico normais, mas referiu deterioração visual nos últimos 2 anos, especialmente à noite. Uma electrorretinografia repetida aos 9 anos de idade revelou uma disfunção retiniana generalizada que afectava tanto o sistema de bastonetes como o de cones, principalmente na interface entre o fotorreceptor e o epitélio pigmentar da retina. A massa ocular aos 6 anos de idade era de 45, 70 e 110 mm para a distância cantal interna, pupilar e cantal externa, respetivamente.
Achados audiológicos: Devido à surdez bilateral severa, o paciente recebeu implantes cocleares aos 4 anos de idade, que foram revistos aos 6 anos e novamente aos 8 anos.
Desenvolvimento escolar: O paciente frequenta uma escola regular e recebe apoio especial para os seus défices visuais e auditivos. Tem um certo atraso de desenvolvimento e frequenta uma turma em que tem um atraso de dois anos em relação aos seus colegas. No entanto, está a fazer bons progressos nesta turma e presume-se que uma grande parte do seu atraso de desenvolvimento se deve à sua deficiência bissensorial e às lacunas na sua escolaridade devido a hospitalizações frequentes.
Confirmação diagnóstica: O diagnóstico suspeito de DBS foi confirmado por testes genéticos, nos quais foi detectada, por sequenciação direta, uma deleção homozigótica de 4 pb (c.11469_11472delTTTG) no exão 60 do gene LRP2.
Literatura:
- “Donnai-Barrow syndrome”, www.orpha.net,(último acesso em 27/09/2024).
- Kantarci S, et al: Síndrome de Donnai-Barrow (DBS/FOAR) numa criança com uma mutação homozigótica LRP2 devido a uma isodisomia completa do cromossoma 2 paterno. Am J Med Genet A 2008; 146A(14): 1842-1847.
- Longoni M, et al: Síndrome de Donnai-Barrow. [Updated 2018 Nov 21] 2008 Aug 28 . In: Adam MP, et al. (Eds). [Internet]GeneReviews® . Seattle (WA): Universidade de Washington, Seattle; 1993-2024.
GP PRACTICE 2024; 19(10): 24-25