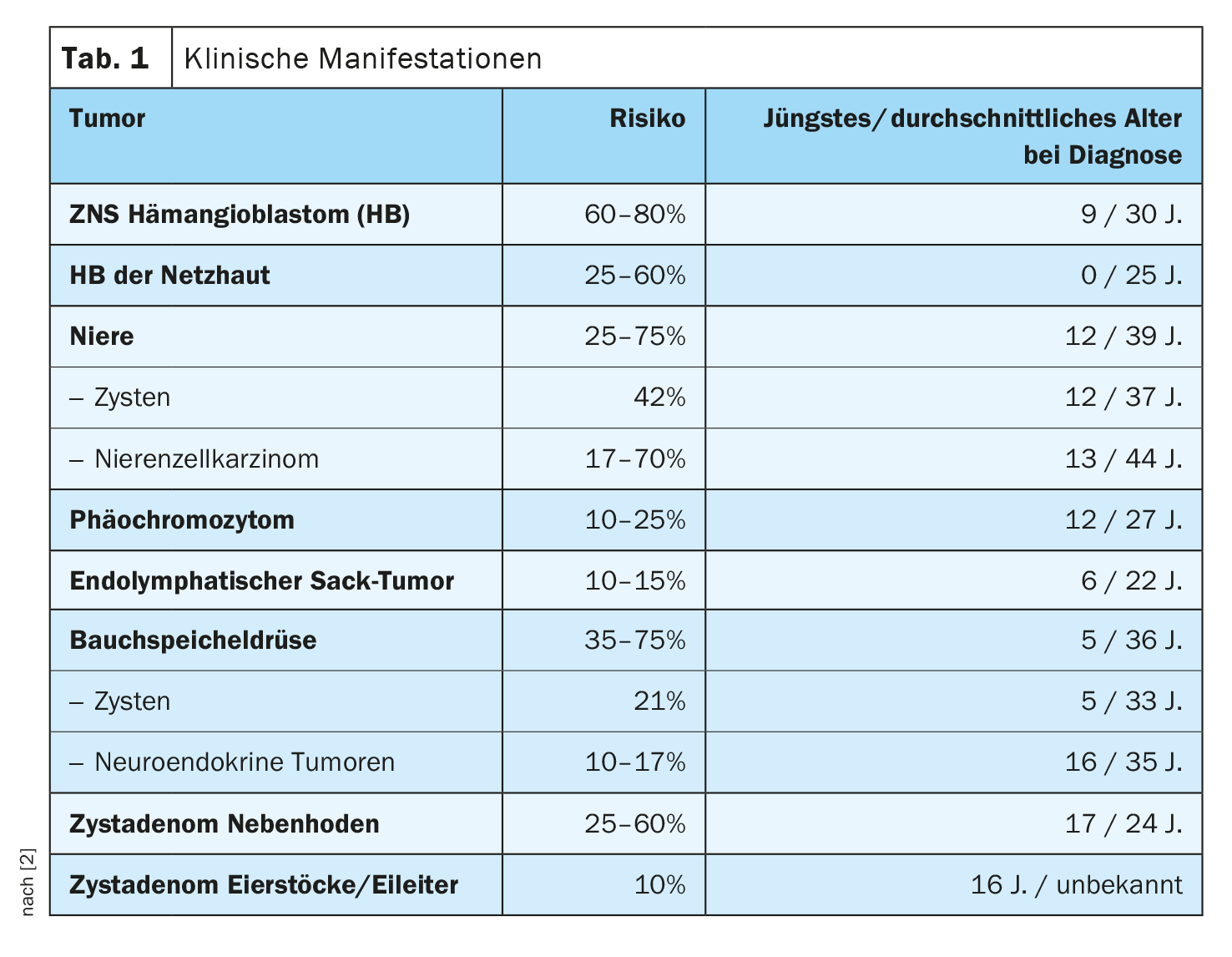
A síndrome de Von Hippel-Lindau é devida a mutações no gene da BVS e caracteriza-se por hemangioblastomas do cérebro, medula espinal e retina do olho. Além disso, os indivíduos afectados têm um risco aumentado de carcinoma de células renais e cistos renais, feocromocitomas, cistos e tumores neuroendócrinos do pâncreas, tumores do saco endolinfático e cistos do epidídimo, bem como dos ovários e trompas de falópio.
Os sintomas dependem do tamanho e da localização dos tumores [1]. As crianças podem sofrer de dores de cabeça e sentir-se tontas ou fracas. Além disso, podem ocorrer distúrbios visuais, possivelmente levando à perda de visão em tumores crescentes da retina, e a tensão arterial elevada. Pode haver uma perda de coordenação. Cerca de 10% das crianças afectadas têm um tumor no ouvido interno, que pode afectar a audição. Sem tratamento, os doentes podem ficar cegos, sofrer danos cerebrais ou morrer. As mortes são geralmente o resultado de complicações de angiomas cerebrais ou cancro do rim.

O diagnóstico precoce é de grande importância. Fenotipicamente, é feita uma distinção entre diferentes tipos de síndrome de Von Hippel-Lindau [1]: O tipo I é caracterizado pela ocorrência de hemangiomas na retina e/ou sistema nervoso central, carcinomas de células renais e/ou tumores neuroendócrinos. No entanto, o risco de feocromocitoma é muito baixo. O Tipo I está associado a mutações sem sentido ou supressões de segmentos genéticos maiores. Em contraste, o tipo II está muitas vezes associado a mutações de missense e o risco de desenvolvimento de feocromocitomas é muito elevado.
Critérios de diagnóstico
O diagnóstico da síndrome de Von Hippel-Lindau é considerado confirmado se for detectada uma mutação no gene da BVS e/ou [2]:
Sem a síndrome de Von Hippel-Lindau na família, na presença de pelo menos 2 das seguintes descobertas:
≥2 hemangioblastomas da retina, medula espinal ou cérebro, ou um único hemangioblastoma juntamente com uma manifestação no abdómen (por exemplo, múltiplos quistos dos rins ou pâncreas).
– Carcinoma das células renais
– Faeocromocitoma
– ELST, cistadenoma dos ovários ou trompas/epidimios ou tumores neuroendócrinos do pâncreas
Com a síndrome de Von Hippel-Lindau na família, pelo menos na presença de. 1 das seguintes descobertas:
– Hemangioblastoma da retina
– Hemangioblastoma da medula espinal ou do cerebelo
– Faeocromocitoma
– Carcinoma das células renais
– Cistos múltiplos dos rins ou do pâncreas
Literatura:
- Institut für Klinische Genetik, Universitätsklinikum Carl Gustav Carus Dresden, www.uniklinikum-dresden.de, (último acesso 17.01.2023)
- Medizinische Hochschule Hannover, www.krebs-praedisposition.de/fuer-patienten-und-familien/von-hippel-lindau-syndrom/#diagnose, (último acesso 17.01.2023)
- Baumgartner-Parzer S: J Klin Endokrinol Stoffw 2020; 13: 37–40.
HAUSARZT PRAXIS 2023; 18(1): 47
InFo ONKOLOGIE & HÄMATOLOGIE 2023; 11(1): 32