La maladie de Flegel est une génodermatose qui se manifeste au cours de la seconde moitié de la vie. Actuellement, le traitement repose sur des produits topiques externes, tels que des préparations contenant de l’urée ou de la cortisone. Afin de déterminer la cause génétique de cette dyskératose rare, une équipe de chercheurs allemands et suisses a mené une étude en utilisant le séquençage de nouvelle génération. Les résultats d’analyse fournissent des informations précieuses pour le développement de nouvelles approches thérapeutiques.
En 1958, le dermatologue allemand Heinz Flegel a été le premier à décrire une nouvelle maladie cutanée rare, caractérisée par de multiples papules plates, asymptomatiques, hyperkératosiques, de couleur brun rougeâtre, situées sur la face d’extension des membres, en particulier au niveau des membres inférieurs et du dos des pieds [1]. Cette pathologie est appelée hyperkératose lenticulaire perstans (HLP) ou maladie de Flegel. En règle générale, les symptômes cutanés apparaissent pour la première fois entre la quatrième et la cinquième décennie de la vie. De nombreuses questions restent en suspens concernant l’étiologie du HLP. Une cause génétique a été suspectée depuis longtemps, mais jusqu’à présent, aucune variante génétique n’a pu être clairement attribuée à l’HLP.
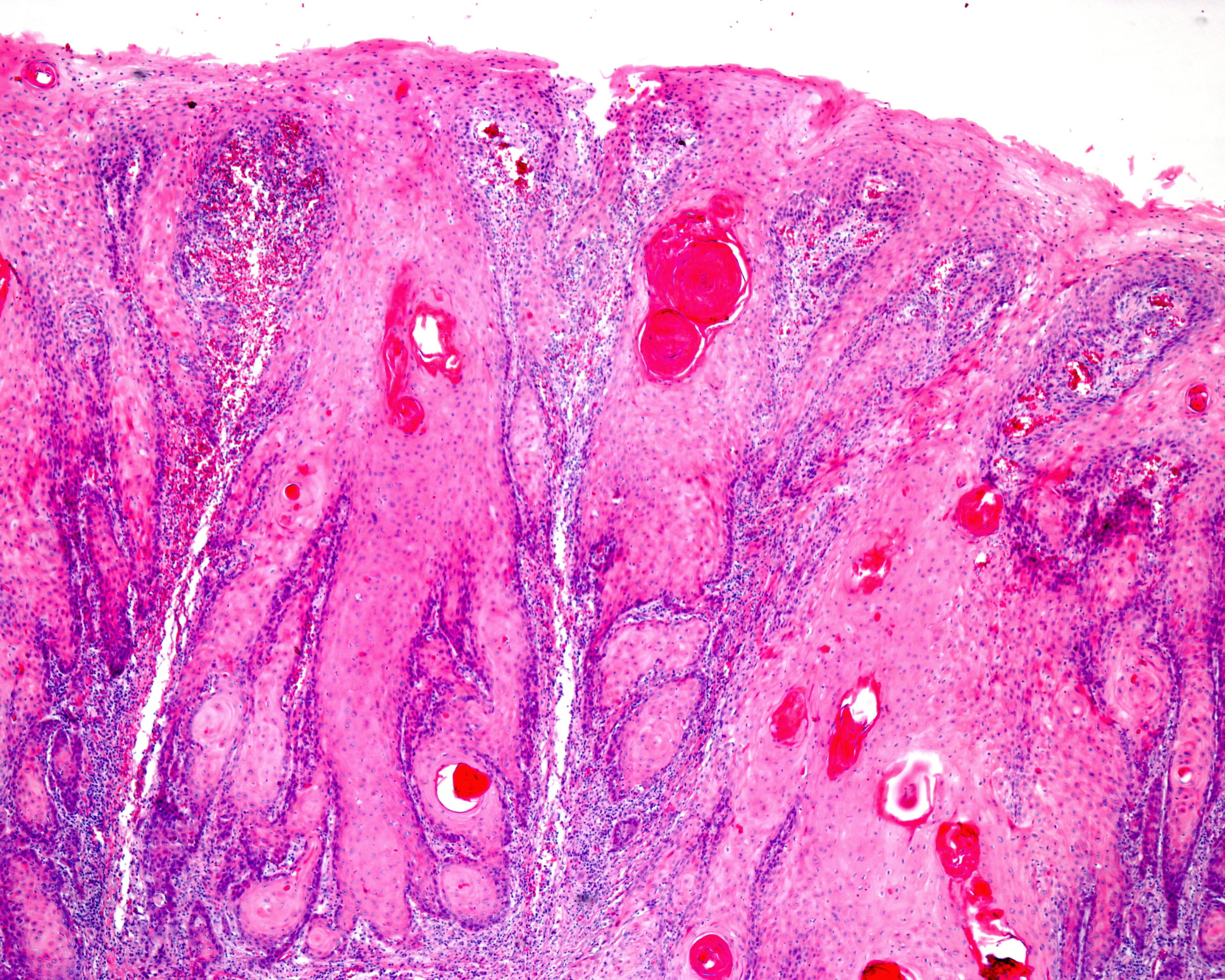
| Vignette de cas Une femme caucasienne de 72 ans s’est présentée pour plusieurs petites papules kératosiques rougeâtres et brunâtres. Les papules sont d’abord apparues sur les membres inférieurs et se sont ensuite étendues aux membres supérieurs et au tronc. (Fig. 1A et B). Il n’y avait pas d’antécédents de maladies cutanées ou systémiques antérieures, à l’exception d’un carcinome épidermoïde cutané bien différencié in situ sur la jambe, qui avait été enlevé deux ans auparavant. Les résultats de la dermatoscopie sont présentés dans la figure 2. L’histopathologie d’une lésion du tronc a montré une hyperkératose lamellaire compacte et épaisse avec parakératose focale sur l’épiderme atrophié, associée à un infiltrat lichénoïde dense de petits lymphocytes dans le derme superficiel avec une accentuation focale dans les zones papillomateuses. Les analyses immunohistochimiques ont révélé des niveaux similaires de lymphocytes T CD4 et CD8, ainsi qu’une faible présence ou une absence de cellules CD20. Après la corrélation clinico-pathologique, le diagnostic final d’hyperkératose lenticulaire généralisée perstans (maladie de Flegel) a été posé. |

Variants “perdus de vue” identifiés dans le gène SPTLC1
Afin de déterminer la cause génétique de l’HLP, les chercheurs ont analysé rétrospectivement le sang EDTA**, les biopsies de peau et les tissus fixés au formol et inclus dans la paraffine (FFPE) de cinq patients en utilisant le séquençage de nouvelle génération (NGS) [1,2]. Chez tous les patients, différentes variantes germinales rares et hétérozygotes ont été identifiées dans le gène SPTLC1 (Serine Palmitoyltransferase Long Chain Base Subunit 1). Il est intéressant de noter que ces variants “perdus de vue” étaient soit de petites délétions, soit des variants d’épissage. En effet, les colorations par immunofluorescence des coupes de peau ont montré une coloration réduite de SPTLC1 chez les patients atteints de HLP.
** Sang rendu non coagulable par l’agent chélatant éthylène diamine tétraacétate (EDTA) en vue d’un examen ultérieur en médecine de laboratoire.

De plus, des preuves d’un “second hit” ont été trouvées dans les lésions affectées sous la forme d’une “perte d’hétérozygosité”. Sur la base de ces résultats, les auteurs de l’étude ont conclu que des variantes germinales pathogènes du gène SPTLC1, combinées à une “perte d’hétérozygotie” dans la peau lésionnelle, étaient responsables de l’apparition du HLP. Il est probable que cela entraîne une diminution de la protéine SPTLC1 – une enzyme clé de la biosynthèse des sphingolipides – et, par conséquent, une perturbation de la voie de signalisation des sphingolipides. L’élucidation des causes génétiques de la maladie de Flegel constitue une base importante pour le développement de nouvelles approches thérapeutiques pour cette maladie cutanée rare, affirment les auteurs [1,2].
Littérature :
- Jägle S, et al. : Des variantes pathogènes du gène SPTLC1 sont à l’origine de la maladie de Flegel (hyperkeratosis lenticularis perstans). FV02/10e Congrès de la DDG, 26-29.04.2023
- Jägle S, et al : Des variantes pathogènes dans le gène SPTLC1 provoquent une hyperkératose lenticulaire perstans. Br J Dermatol 2023 ; 188(1) : 94-99.
- Stabile G, et al. : Hyperkératose lenticulaire perstans (maladie de Flegel) étendue et durable : Caractéristiques clinico-pathologiques et dermoscopiques d’une présentation rare. Dermatopathology 2023 ; 10(1) : 46-51. www.mdpi.com/2296-3529/10/1/6,(dernière consultation 18.08.2023).
DERMATOLOGIE PRATIQUE 2023 ; 33(4) : 40