La cardiomyopathie hypertrophique (CMH) est la cardiomyopathie génétique la plus fréquente au monde et touche environ 1 personne sur 500. Les interventions thérapeutiques actuelles comprennent l’optimisation du mode de vie, les médicaments, les thérapies de réduction du septum et, rarement, la transplantation cardiaque. Les progrès dans la compréhension des variantes génétiques responsables de la maladie dans la CMH et de leurs mécanismes moléculaires ont ouvert la voie à des thérapies ciblées et à la mise en œuvre d’une médecine de précision et personnalisée. Les résultats des recherches précliniques sont prometteurs et soulèvent la question de savoir si une guérison de certains sous-types de CMH pourrait être possible à l’avenir.
La cardiomyopathie hypertrophique (CMH) est une maladie myocardique primaire caractérisée par une hypertrophie inexpliquée du ventricule gauche et survenant en l’absence de conditions de charge anormales telles que l’hypertension ou la sténose aortique. La présentation clinique de la CMH est très variable, allant d’individus asymptomatiques à ceux présentant des symptômes graves tels que l’insuffisance cardiaque, l’arythmie et la mort subite d’origine cardiaque (MSC). La base génétique de la CMH est également hétérogène, avec des mutations dans au moins huit gènes de sarcomères identifiés comme pathogènes et une proportion importante de cas associés à des variants génétiques de signification indéterminée (VUS).
Paysage thérapeutique actuel pour l’HCM
Mesures relatives au mode de vie : L’optimisation du mode de vie est essentielle pour la prise en charge de la CMH. Éviter l’hypertension non contrôlée et l’obésité est essentiel pour prévenir l’aggravation du phénotype de la CMH. Des études récentes ont également mis en évidence les avantages d’une activité physique régulière, légère à modérée, dans l’amélioration de la qualité de vie et des résultats cardiovasculaires des patients atteints de CMH. Auparavant, les patients atteints de CMH étaient mis en garde contre une activité physique intense, mais des recherches récentes suggèrent que l’exercice modéré peut être sans danger et bénéfique.
Traitement médicamenteux : des médicaments tels que les bêtabloquants et les inhibiteurs des canaux calciques sont souvent utilisés pour soulager les symptômes et réduire le risque de complications de la CMH. Ces médicaments agissent en diminuant la fréquence cardiaque et en réduisant la contractilité du cœur, ce qui contribue à réduire l’obstruction de la voie de sortie du ventricule gauche. Des traitements plus récents, comme le mavacamten, un inhibiteur de la myosine, ont montré des résultats prometteurs dans des études récentes et ont amélioré les symptômes subjectifs et objectifs chez les patients atteints de CMH obstructive. Le mavacamten modifie directement les chaînes lourdes de la β-myosine afin de réduire l’affinité entre l’actine et la myosine et de normaliser ainsi la fonction cardiaque.
Les thérapies interventionnelles : Les procédures interventionnelles, y compris la myectomie du septum et l’ablation du septum d’alcool, sont effectuées pour éliminer l’obstruction de l’orifice de sortie du ventricule gauche (OVG). La myectomie septale est une intervention chirurgicale qui consiste à retirer une partie du septum épaissi afin d’améliorer le flux sanguin du ventricule gauche. Cette méthode est typiquement utilisée chez les patients jeunes. La septumablation à l’alcool, une alternative moins invasive, est plus souvent pratiquée chez les patients plus âgés. Elle consiste à injecter de l’alcool dans les artères septales afin de provoquer un infarctus contrôlé et de réduire l’hypertrophie. Malgré des taux plus élevés d’arythmie et de cicatrices par rapport à la myectomie septale, l’ablation septale à l’alcool peut donner d’excellents résultats dans les centres expérimentés.
Prévention de la mort subite d’origine cardiaque : les défibrillateurs cardioverteurs implantables (DAI) sont recommandés pour les personnes présentant un risque élevé de MCS. Les progrès réalisés dans la stratification des risques et la disponibilité des DAI ont permis de réduire considérablement la mortalité liée à la MHC. L’évaluation du risque se fait à l’aide de calculateurs de risque basés sur des lignes directrices, qui prennent en compte des facteurs tels que l’épaisseur du septum, la présence de tachycardies ventriculaires non soutenues et les antécédents familiaux.
Transplantation cardiaque : dans de rares cas, lorsque les patients ne répondent pas à tous les autres traitements et développent une insuffisance cardiaque avancée, une transplantation cardiaque peut être nécessaire. Ces patients représentent environ 1,6% de ceux qui sont en attente d’une transplantation cardiaque aux États-Unis. Malgré leur rareté, le taux de survie après une transplantation cardiaque chez les patients atteints de CMH est similaire à celui des patients atteints d’autres cardiomyopathies.
Thérapie génique – la nouvelle frontière ?
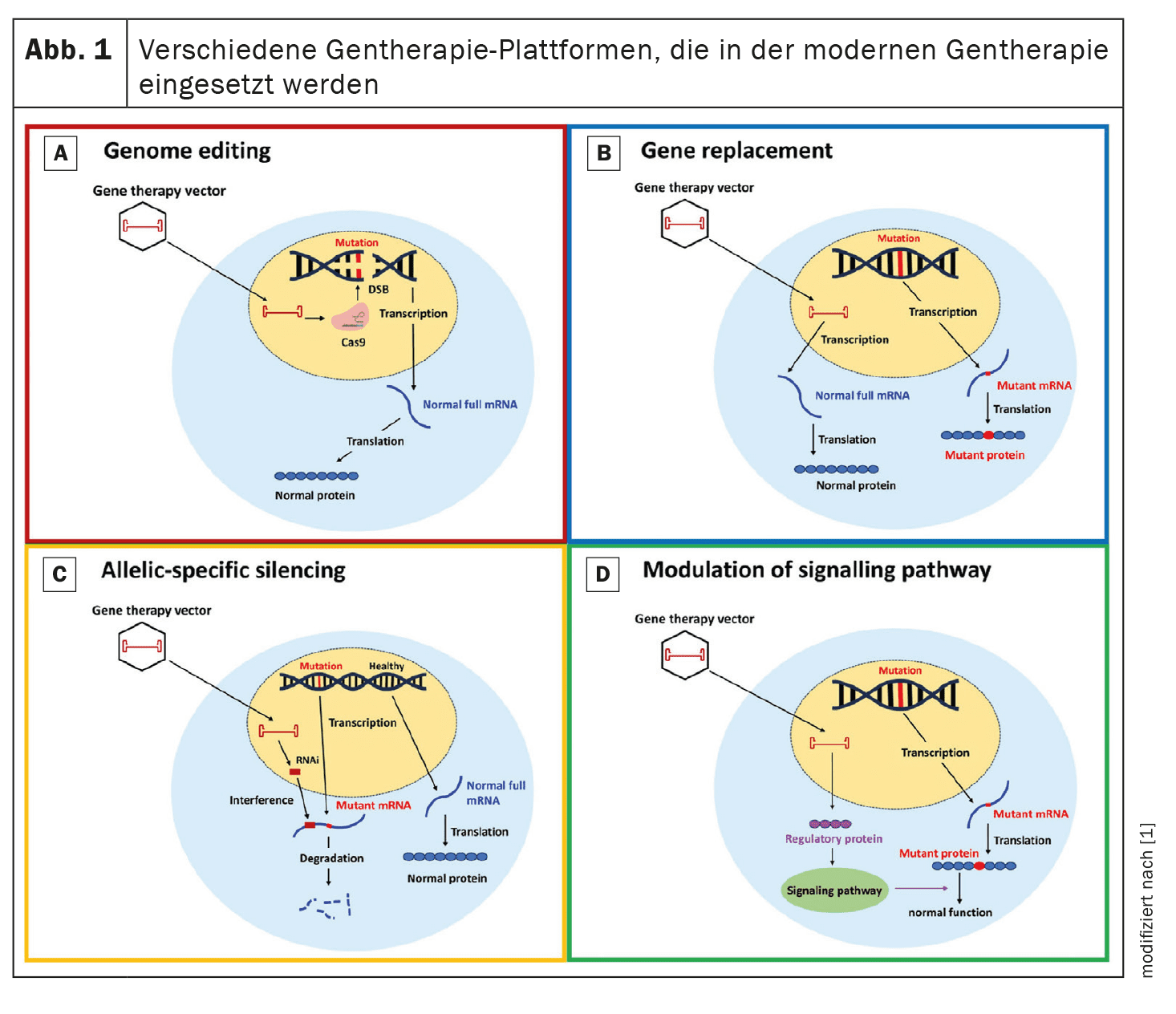
La thérapie génique vise à corriger ou à atténuer les mutations génétiques responsables de la CMH. Quatre approches principales sont examinées dans cette revue : L’édition génomique, le remplacement de gènes, le silencing spécifique d’allèle et la modulation des voies de signalisation.
Édition du génome : L’édition du génome à l’aide de la technologie CRISPR/Cas9 a démontré son potentiel pour corriger les mutations génétiques associées à la HCM dans des modèles précliniques.
CRISPR/Cas9 utilise une nucléase programmable qui génère des cassures double brin d’ADN ciblées qui peuvent être réparées par jointure finale non homologue (NHEJ) ou par recombinaison homologue (HDR).
Cependant, des défis tels que les effets hors cible et la nécessité de méthodes de livraison précises restent des obstacles importants.
Les recherches se concentrent sur l’amélioration de la spécificité et de l’efficacité de cette technologie afin de minimiser les modifications génétiques involontaires.
Remplacement de gène : la thérapie de remplacement de gène implique l’introduction d’une copie fonctionnelle du gène muté.
Cette approche est particulièrement pertinente pour les mutations conduisant à l’haploinsuffisance.
Les études en cours, telles que l’utilisation de vecteurs de virus adéno-associés (AAV), visent à évaluer l’efficacité et la sécurité de cette stratégie chez les patients atteints de CMH.
Par exemple, le remplacement du gène MYBPC3, qui est souvent muté dans la CMH, fait actuellement l’objet d’essais cliniques.
Dans les études précliniques, le remplacement du gène défectueux a permis d’améliorer la fonction myocardique et de prévenir les phénotypes de la CMH.
Silencing spécifique à l’allèle : Le silencing spécifique à l’allèle vise à supprimer l’allèle muté tout en préservant la fonction de l’allèle normal. Cette approche utilise de petits ARN interférents (siRNA) pour dégrader sélectivement l’ARNm muté et réduire la production de protéines anormales. Cette approche est particulièrement utile dans le cas de mutations dominantes où la protéine mutée a un effet délétère. Des études précliniques ont montré que les siRNA peuvent efficacement réduire l’expression de l’allèle muté, ce qui entraîne une amélioration de la fonction cardiaque et une réduction de l’hypertrophie.
Modulation des voies de signalisation : La modulation des voies de signalisation clés impliquées dans la pathogenèse de la CMH offre une autre approche thérapeutique. Par exemple, l’augmentation de l’expression de SERCA2a, une protéine impliquée dans l’équilibre calcique, a montré un potentiel de réduction de l’hypertrophie et d’amélioration de la fonction cardiaque dans des modèles précliniques. Une autre approche consiste à moduler la phosphorylation de la chaîne légère régulatrice de la myosine (myosine RLC) afin d’améliorer la fonction contractile du cœur.
Défis et orientations futures
Malgré des progrès prometteurs, plusieurs défis entravent la mise en œuvre clinique des thérapies géniques pour la CMH. Il s’agit notamment de
- Préoccupations en matière de sécurité : les risques liés aux vecteurs AAV, tels que les réactions immunitaires et les effets hors cible, doivent être abordés de manière approfondie. Les récents décès liés aux thérapies géniques basées sur les AAV rappellent les dangers potentiels et la nécessité de continuer à améliorer la sécurité de ces technologies.
- Méthodes de livraison : la livraison efficace et ciblée de thérapies géniques au tissu cardiaque reste un défi technique. Le développement de vecteurs avec un tropisme cardiaque spécifique et amélioré pourrait aider à minimiser les transductions hors cible et à réduire les doses nécessaires.
- Hétérogénéité génétique : la diversité génétique de la CMH rend difficile le développement d’approches universelles de thérapie génique. La HCM étant causée par une multitude de mutations, le développement d’ARNg spécifiques pour chaque mutation et la limitation par des séquences PAM nécessitent des recherches et une optimisation supplémentaires.
- Éthique et accès équitable : il est essentiel de s’assurer que les thérapies géniques sont accessibles à tous les patients, quel que soit leur statut socio-économique. Une distribution équitable de ces traitements avancés doit être une priorité afin d’éviter les inégalités en matière de santé.
Conclusion
Les progrès de la thérapie génique sont très prometteurs pour le traitement futur de la CMH. Alors que les recherches précliniques ont montré un potentiel, la mise en œuvre de ces thérapies dans la pratique clinique nécessitera des défis scientifiques, techniques et éthiques considérables. Un accès équitable à ces traitements avancés doit être garanti afin de réaliser leur plein potentiel d’amélioration des résultats pour les patients atteints de CMH. La poursuite de la recherche et du développement de thérapies géniques sûres et efficaces pourrait révolutionner la manière dont la CMH et les autres cardiomyopathies génétiques sont traitées.
Source :
- Paratz ED, Mundisugih J, Rowe SJ, et al.: Gene Therapy in Cardiology: Is a Cure for Hypertrophic Cardiomyopathy on the Horizon? Can J Cardiol 2024 May; 40(5): 777–788. doi: 10.1016/j.cjca.2023.11.024. Epub 2023 Nov 25. PMID: 38013066.
CARDIOVASC 2024; 23(2): 24–25