Le malattie polmonari interstiziali sono frequentemente riscontrate dai medici di base nella pratica, soprattutto nelle forme iniziali. La differenziazione tra le varie forme spesso non è evidente a prima vista e richiede una diagnosi precisa con l’anamnesi. Un esperto dell’Ospedale Universitario di Basilea ha fornito una panoramica delle pneumopatie interstiziali da considerare.
La fibrosi polmonare è una malattia relativamente rara associata a una compressione del parenchima polmonare. Questo cambiamento porta a una riduzione del volume polmonare e a un’ostruzione nell’assorbimento di ossigeno. Dal punto di vista prognostico, il rimodellamento fibrotico del polmone deve essere considerato sfavorevole, ha dichiarato la dottoressa Kathleen Jahn della Clinica di Pneumologia dell’Ospedale Universitario di Basilea.
Fibrosi polmonare idiopatica
La fibrosi polmonare idiopatica ha una prognosi molto sfavorevole, con un tempo di sopravvivenza mediano di 1,5-3 anni dopo la diagnosi iniziale. Tuttavia, ci possono essere decorsi fulminanti, ricadute, ma anche progressioni relativamente lente.
Dal punto di vista fisiopatologico, si tratta di una reazione infiammatoria in cui vengono rilasciate citochine e si accumulano prodotti infiammatori. Lo stress ossidativo aumenta, portando a una compattazione della matrice extracellulare. I fibroblasti sono attivati e c’è una disregolazione degli ormoni della crescita nel tessuto polmonare.
Nella funzione polmonare, questo si presenta come un restringimento del polmone. “Diventa più piccola e più solida e non può più espandersi. Nella pratica clinica quotidiana, si nota che il torace non può più muoversi così bene nella fibrosi polmonare avanzata, ci sono delle restrizioni”, ha spiegato il dottor Jahn. La capacità di un secondo (flusso di picco) non deve necessariamente essere limitata, ma il volume totale si riduce complessivamente e i polmoni non possono più riempirsi completamente.
Bronchiolite respiratoria (RB-ILD)
La RB-ILD è la più innocua delle pneumopatie interstiziali. I pazienti hanno in genere una storia di fumo persistente da molti anni (almeno un anno). 20 PY), e la maggior parte di loro non sta cercando di interrompere la nicotina a lungo termine quando si presentano con la bronchiolite. I sintomi sono aspecifici: tosse (produttiva e non produttiva) e difficoltà respiratorie nello sforzo, auscultando di solito rantoli. La TAC mostra un modello centrinodulare (OL accentuato), piccoli noduli (<5 mm) si trovano in tutto il parenchima polmonare, alcuni dei quali sono circondati da vetro lattiginoso, può esserci un ispessimento incipiente della parete bronchiale. Il classico rimodellamento fibrotico si osserva molto raramente nella RB-ILD.
Il gold standard nella diagnostica è la broncoscopia con lavaggio (BAL). I macrofagi dei fumatori sono i più visibili. Meno comuni e solitamente non necessarie sono le biopsie transbronchiali (TBB), che mostrano depositi di antracosi bronchiolocentrica e infiammazione neutrofila. La terapia è relativamente semplice: il primo passo è smettere di fumare. Due terzi dei casi mostrano un miglioramento clinico se riescono a smettere di fumare in modo costante. Il prednisone (0,5 mg/kgKG iniziali) è raramente necessario e solo in caso di presentazione avanzata.
Sarcoidosi
L’età media di manifestazione della sarcoidosi è compresa tra 20 e 60 anni. Anche in questo caso, i sintomi sono aspecifici: tosse improduttiva, intolleranza alle prestazioni, dolore al petto e febbre sono tra questi, ma anche costellazioni poco chiare di infezioni. Meno frequentemente, possono verificarsi anche perdita di peso e affaticamento.
La diagnostica per immagini (TAC del torace) mostra una linfoadenopatia bifilare mediastinica nel 50%, possono comparire anche noduli a vetro di latte e, negli stadi avanzati con infiammazione cronica, possono verificarsi anche distruzione parenchimale e cambiamenti fibrotici. Inoltre, una PET-CT può essere utile per rilevare la linfoadenopatia e per escludere diagnosi differenziali di natura maligna. Nelle fasi iniziali, la diagnosi di solito può essere evidenziata solo dalla spiroergometria. Sotto stress, si può osservare un calo rilevante della pO2, che può essere un segno precoce di sarcoidosi.
La broncoscopia è di nuovo il gold standard per confermare la diagnosi. Si riscontra una linfocitosi media (circa 20-30%) con un aumento del quoziente CD4/CD8 >2 nel BAL. Un rilievo simile a un ciottolo indica un’infiammazione granulomatosa (SH-Bx), e al momento della rimozione della biopsia (TBB) si trovano granulomi produttivi, non caseosi. La terapia viene determinata individualmente a seconda del coinvolgimento dell’organo e della clinica:
I pazienti asintomatici e quelli con marcatori predittivi negativi non richiedono la terapia.
Le indicazioni per la terapia sono l’ipercalcemia/ipercalciuria, il coinvolgimento occulto e le limitazioni della diffusione.
Di solito si inizia con una monoterapia di prednisone 0,3-0,6 mg/kgKG (circa 20-40 mg/giorno). Questo viene applicato con incrementi di 5-10 mg ogni 2-4 settimane. La durata iniziale della terapia deve essere di circa 3-6 mesi. Se questo non porta al miglioramento desiderato, si può aggiungere l’azatioprina o il micofenolato. Questo colpisce soprattutto i pazienti con diabete di tipo 2 o osteoporosi. Come sempre con la terapia steroidea cronica, bisogna anche ricordare di stabilire una protezione gastrica (PPI) e di eseguire la profilassi della pneumocystis (3×/settimana), ha ricordato l’esperto. È anche molto importante non aggiungere calcio nella sarcoidosi, poiché questo porta all’attivazione dei macrofagi.
OP e COP
La polmonite organizzativa (OP) e la polmonite organizzativa criptogenetica (COP) hanno lo stesso aspetto alla TAC. In caso di intervento chirurgico, di solito sono presenti cause infettive classiche. “Al contrario, descriviamo come criptogenetiche solo quelle polmoniti che non hanno un chiaro fattore scatenante e che quindi sono considerate idiopatiche”, afferma il dottor Jahn.
In OP e COP, c’è un danno all’epitelio alveolare e un’essudazione di fibrina nello spazio alveolare. I cosiddetti infiltrati migranti sono tipici della COP. Dal punto di vista terapeutico, l’intervento chirurgico può essere eseguito con un regime di strategia watch-and-wait se la limitazione funzionale è solo lieve. Il prednisone (PDN) è nuovamente indicato per i casi più gravi. “Vede: Il cortisone è un farmaco relativamente miracoloso nelle pneumopatie interstiziali”. Tuttavia, a causa di un tasso di ricaduta di circa il 30%, anche in questo caso si dovrebbe scendere lentamente.
Se c’è una ricaduta, ricominci con dosi più elevate di steroidi. Le possibili diagnosi differenziali in caso di frequenti ricadute nonostante una buona risposta alla terapia PDN consolidata includono una sindrome da immunodeficienza, la granulomatosi con poliangite e altre malattie reumatologiche, la tiroidite cronica, ma anche le malattie infiammatorie croniche intestinali.
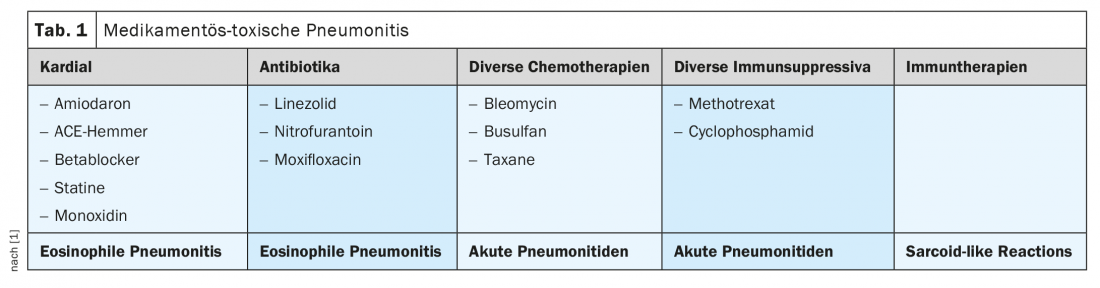
Polmonite tossica da farmaci
In caso di polmonite tossica da farmaci (Tab. 1) con vari fattori scatenanti, il BAL è solitamente utile per confermare la diagnosi; raramente è necessaria la TBB. Dal punto di vista terapeutico, è indicata l’interruzione dell’agente causale; a seconda della clinica, può essere appropriata una terapia di supporto della PDN.
Fibrosi polmonare idiopatica (IPF)
La maggior parte delle pneumopatie interstiziali risponde molto bene agli steroidi – l’IPF, invece, è completamente resistente agli steroidi. Tuttavia, da alcuni anni sono disponibili opzioni terapeutiche mirate.
L’IPF è spesso familiare, soprattutto se la persona colpita ha meno di 55 anni . Mostra il classico quadro di una restrizione, i volumi diventano più piccoli, ci sono modelli di iperventilazione. In laboratorio, si deve escludere una costellazione reumatica come malattia di base. Meno sono i cambiamenti tipici che si possono vedere sulla TAC, meno è probabile l’IPF, ha spiegato l’esperto.
I due principi attivi pirfenidone e nintedanib possono essere utilizzati per il trattamento. Entrambi hanno effetti antifibrotici, antiproliferativi e antinfiammatori. Entrambi sono stati testati in ampi studi e sono sul mercato da diversi anni. Entrambi riducono il declino del volume polmonare. Ma: non provocano un processo che può essere invertito – la fibrosi presente non può essere ridotta di nuovo. Inoltre, entrambi i farmaci sono relativamente poco tollerati e causano, tra l’altro, disturbi addominali, nausea, diarrea e dispepsia. Il pirfenidone provoca un’elevata fototossicità – i pazienti che amano il sole dovrebbero quindi essere adattati a nintedanib, secondo il consiglio del dottor Jahn. Nintedanib, tuttavia, comporta un maggior numero di epatopatie sul versante opposto. È inoltre controindicato in caso di assunzione di anticoagulanti orali, a causa dell’aumento del rischio di sanguinamento.
Messaggi da portare a casa
- Valutazione precoce del sito funzionale e chiarimento funzionale nella dispnea progressiva
- Anamnesi completa per quanto riguarda la familiarità, l’esposizione alle tossine, la polimedicazione.
- Controlli severi
- Molte ILD rispondono agli steroidi e mostrano una buona risposta.
- L’IPF è un caso speciale, in quanto NON risponde agli steroidi, ma la sua progressione può essere ritardata con sostanze antiproliferative.
- Il BAL di solito rimane aspecifico.
- È indicata la diagnosi mediante biopsia polmonare (endoscopica o chirurgica).
- Attenzione agli effetti collaterali dei farmaci
Fonte:
- Jahn K: Malattie polmonari interstiziali comuni – diagnosi e terapia. Aggiornamento FomF Medicina Interna Generale, 25.1.2022 (virtuale)
PRATICA GP 2022; 17(5): 43-44
InFo PNEUMOLOGIA & ALLERGOLOGIA 2022; 4(2): 38-39