L’ipertensione polmonare è suddivisa in 5 gruppi diversi, con il gruppo 1 che riflette l’ipertensione arteriosa polmonare (PAH). Le terapie farmacologiche specifiche per la PH approvate finora interferiscono con tre diverse vie di segnalazione (via di segnalazione dell’NO, via di segnalazione della prostaciclina e via di segnalazione del recettore dell’endotelina). Sono successe molte cose da quando il primo farmaco specifico per la PAH (bosentan) è stato approvato in Europa circa 20 anni fa. Sono state aggiunte numerose altre sostanze che hanno migliorato la prognosi dei pazienti affetti da PAH.
L’ipertensione polmonare è suddivisa in 5 gruppi diversi, con il gruppo 1 che riflette l’ipertensione arteriosa polmonare (PAH). Le terapie farmacologiche specifiche per la PH approvate finora interferiscono con tre diverse vie di segnalazione (via di segnalazione dell’NO, via di segnalazione della prostaciclina e via di segnalazione del recettore dell’endotelina). Sono successe molte cose da quando il primo farmaco specifico per la PAH (bosentan) è stato approvato in Europa circa 20 anni fa. Sono state aggiunte numerose altre sostanze che hanno migliorato la prognosi dei pazienti affetti da PAH.
Mentre per molto tempo la PAH idiopatica (IPAH) è stata diagnosticata principalmente in pazienti giovani senza comorbidità clinicamente rilevanti, negli ultimi anni è emerso un cambiamento demografico. Un numero crescente di pazienti anziani con comorbidità polmonari e/o cardiache riceve attualmente la diagnosi di ipertensione polmonare precapillare grave e viene classificato come paziente IPAH secondo la classificazione attuale. Diverse analisi di database [1,2] hanno dimostrato che i pazienti con IPAH hanno caratteristiche patologiche molto diverse e che esistono diversi cluster o fenotipi. Va sottolineato che la capacità di diffusione (DLCO) è, tra l’altro, un fattore discriminatorio e prognostico cruciale [3–5].
Questo documento riproduce la presentazione “PAH: cosa c’è oltre la nuova linea guida”, tenuta in occasione del 63° Congresso tedesco degli pneumologi a Düsseldorf il 30.03.2023. La presentazione ha mostrato i dati dello studio di pazienti con diagnosi di IPAH che presentavano lievi alterazioni dell’impalcatura polmonare alla TAC del torace e/o un grave difetto di diffusione alveolocapillare di eziologia non chiara.
Le comorbilità come fattore prognostico – situazione attuale dello studio
Già nel 2016, in occasione della Conferenza di Consenso di Colonia, è stato discusso tra gli esperti di PH di lingua tedesca che la popolazione di pazienti affetti da IPAH inclusi in studi terapeutici prospettici, multicentrici e controllati con placebo, spesso non è comparabile con i pazienti inclusi in vari registri nazionali di PH e dominano anche nella pratica clinica quotidiana. Già all’epoca si notava che questi ultimi erano più anziani, avevano più comorbidità ed erano fenotipicamente simili ai pazienti con malattia cardiaca e/o polmonare sinistra a causa dei loro fattori di rischio, ma venivano classificati come IPAH secondo i criteri di definizione attualmente validi. A quel tempo, sono stati introdotti i termini di pazienti IPAH “tipici” e “atipici” per riflettere questo sviluppo. La “IPAH tipica” era definita emodinamicamente a quel tempo, dopo l’esclusione di tutte le cause note di PH, da una pressione arteriosa polmonare media (mPAP) ≥25 mmHg e una pressione di occlusione arteriosa polmonare (PAWP) ≤15 mmHg. Inoltre, analogamente allo studio AMBITION (ambrisentan più tadalafil rispetto ad ambrisentan o tadalafil in monoterapia), solo un massimo di due fattori di rischio per la disfunzione diastolica ventricolare sinistra (insufficienza cardiaca con frazione di eiezione ventricolare sinistra conservata, HFpEF: ipertensione arteriosa, malattia coronarica, diabete mellito e obesità con BMI >30 kg/m2) e la capacità di diffusione (DLCO) deve essere almeno il 45% del valore nominale. [6,7]. Nel gruppo della “IPAH atipica”, che non differiva nel profilo emodinamico dalla “IPAH classica”, i pazienti erano prevalentemente anziani (per lo più >65 anni) e presentavano il profilo di rischio o le malattie concomitanti dei pazienti con malattia cardiaca o polmonare sinistra. A quel tempo, erano chiamati rispettivamente “pazienti IPAH con fenotipo cardiaco” e “pazienti IPAH con fenotipo polmonare”.
I pazienti con un fenotipo polmonare erano caratterizzati da una tismografia corporea (quasi) normale e da risultati di TAC del torace, spesso da una grave ipossiemia e da una marcata riduzione della capacità di diffusione (DLCO <45% del valore target) (Tab. 1) . Per quanto riguarda la terapia PH mirata, la monoterapia è stata inizialmente raccomandata per i pazienti “atipici”, in quanto l’efficacia e la tollerabilità dei farmaci specifici per la PAH in questa popolazione non erano state studiate a sufficienza per poter formulare raccomandazioni basate sull’evidenza. I dati del registro hanno mostrato che questi pazienti sono stati trattati prevalentemente con inibitori della PDE5 e sono stati anche piuttosto riluttanti ad assumere una terapia combinata durante il corso del trattamento.

DLCO come fattore di rischio prognostico – situazione attuale dello studio
Già nel 2010 è stato pubblicato uno studio [4] che descriveva una correlazione tra DLCO e sopravvivenza nei pazienti con PAH. Qui sono stati elaborati tre intervalli per la DLCO (DLCO >64%, DLCO 43-63%, DLCO <43%) che discriminano bene il rischio di morte. In questo studio, la diminuzione della DLCO è stata associata in modo multivariato all’età del paziente, alla presenza di collagenosi, alla diminuzione della capacità di esercizio funzionale, alla richiesta di ossigeno, alla riduzione dei volumi polmonari e ai cambiamenti morfologici della TAC. Queste correlazioni erano indipendenti dall’emodinamica cardio-polmonare. I pazienti che avevano il range di DLCO più basso (<43%) avevano un rischio di morte aumentato di 2,7 volte.
Nel 2013, Trip et al. [8] che i pazienti con IPAH con una DLCO <45% hanno una prognosi significativamente peggiore rispetto ai pazienti con IPAH con una DLCO superiore corretta per età. Complessivamente, circa il 75% di tutti i pazienti con IPAH presentava una DLCO ridotta (prevalentemente da lieve a moderata). L’analisi ha escluso i pazienti con cause note di grave riduzione della diffusione, come il forame ovale pervio, le collagenosi, la PVOD (malattia veno-occlusiva polmonare), gravi malattie del cuore e del polmone sinistro. Le alterazioni TC lievi e moderate non hanno escluso la diagnosi di IPAH in questo studio.
Trip et al. ha dimostrato che i pazienti con IPAH + DLCO <erano mediamente più anziani del 45% (67 anni contro 49 anni), prevalentemente di sesso maschile, con più probabilità di avere una CHD e più probabilità di essere fumatori. I parametri funzionali polmonari (FEV1, FEV1/FVC e TLC) tendevano ad essere più bassi e le alterazioni morfologiche della TAC (enfisema o fibrosi da lieve a moderata) erano più frequenti rispetto ai pazienti con una DLCO di ≥45%. L’emodinamica polmonare, invece, era paragonabile. Tuttavia, questi pazienti hanno mostrato una minore capacità di recupero funzionale (test del cammino di 6 minuti).
La sopravvivenza a 3 anni (DLCO <45% vs. DLCO ≥45%) è stata rispettivamente del 54% e dell’86%, e a 5 anni del 30% e dell’80%. Gli autori hanno concluso dai loro risultati che la grave riduzione della DLCO era probabilmente dovuta a un sottotipo di PH da considerare separatamente, che potrebbe essere innescato dall’inalazione di fumo di sigaretta.
Nel 2017, il gruppo di ricerca di Hannover guidato da Olsson et al. [5] che i pazienti affetti da IPAH con una funzione polmonare completamente normale e risultati TC del torace non significativi, ma con una DLCO significativamente ridotta (<45% del target) sono prognosticamente paragonabili ai pazienti con CPFE (fibrosi polmonare ed enfisema combinati) . Entrambi i gruppi erano prevalentemente uomini con un’anamnesi da fumatore e non differivano nelle loro caratteristiche, tranne che per i cambiamenti funzionali polmonari e morfologici della TAC.
Tutti i pazienti di questo studio sono stati trattati con inibitori della PDE5, ma sia la distanza di cammino nel test del cammino di 6 minuti che la pressione parziale di ossigeno (paO2) tendevano a peggiorare in modo comparabile con la terapia in entrambi i gruppi. La mortalità in entrambi i gruppi non differiva significativamente a 1, 2 e 4 anni (80% / 76% / 38% contro 64% / 42% / 42%). Gli autori hanno concluso che la causa della grave riduzione della DLCO nei loro pazienti con IPAH era probabilmente dovuta alla “vaculopatia polmonare legata al fumo”.
I dati del Registro PH di Sheffield (ASPIRE; Regno Unito) mostrano che i pazienti affetti da IPAH con lievi alterazioni parenchimali nella TAC del torace non migliorano la capacità di esercizio nonostante la terapia combinata specifica per la PAH. Questi pazienti avevano anche una prognosi significativamente peggiore rispetto ai pazienti con IPAH senza anomalie del torace da TAC. Nei pazienti con IPAH senza alterazioni TC ma con una DLCO <45%, la prognosi aggiustata per età in questo studio di registro era anche significativamente peggiore rispetto ai pazienti con IPAH con una DLCO ≥45% [1].
Nel 2020, i pazienti affetti da IPAH nel database europeo COMPERA (Comparative, Prospective Registry of Newly Initiated Therapies for Pulmonary Hypertension) sono stati sottoposti a un’analisi cluster basata sulle variabili età, sesso (maschio vs. femmina), stato di fumatore ([Ex]Fumatori: sì vs. no), DLCO (<45% vs. ≥45% del set point) e la presenza o l’assenza di almeno un fattore di rischio per la disfunzione diastolica ventricolare sinistra (obesità, ipertensione arteriosa, malattia coronarica e diabete mellito). I cluster trovati sono stati analizzati in termini di caratteristiche basali, di risposta alle terapie specifiche per la PH (cambiamenti nella distanza del cammino di 6 minuti, nella classe funzionale e nei livelli ematici dei biomarcatori) e di prognosi, tra gli altri. Sono stati trovati tre diversi cluster (Fig. 1; Cluster 1-3) .
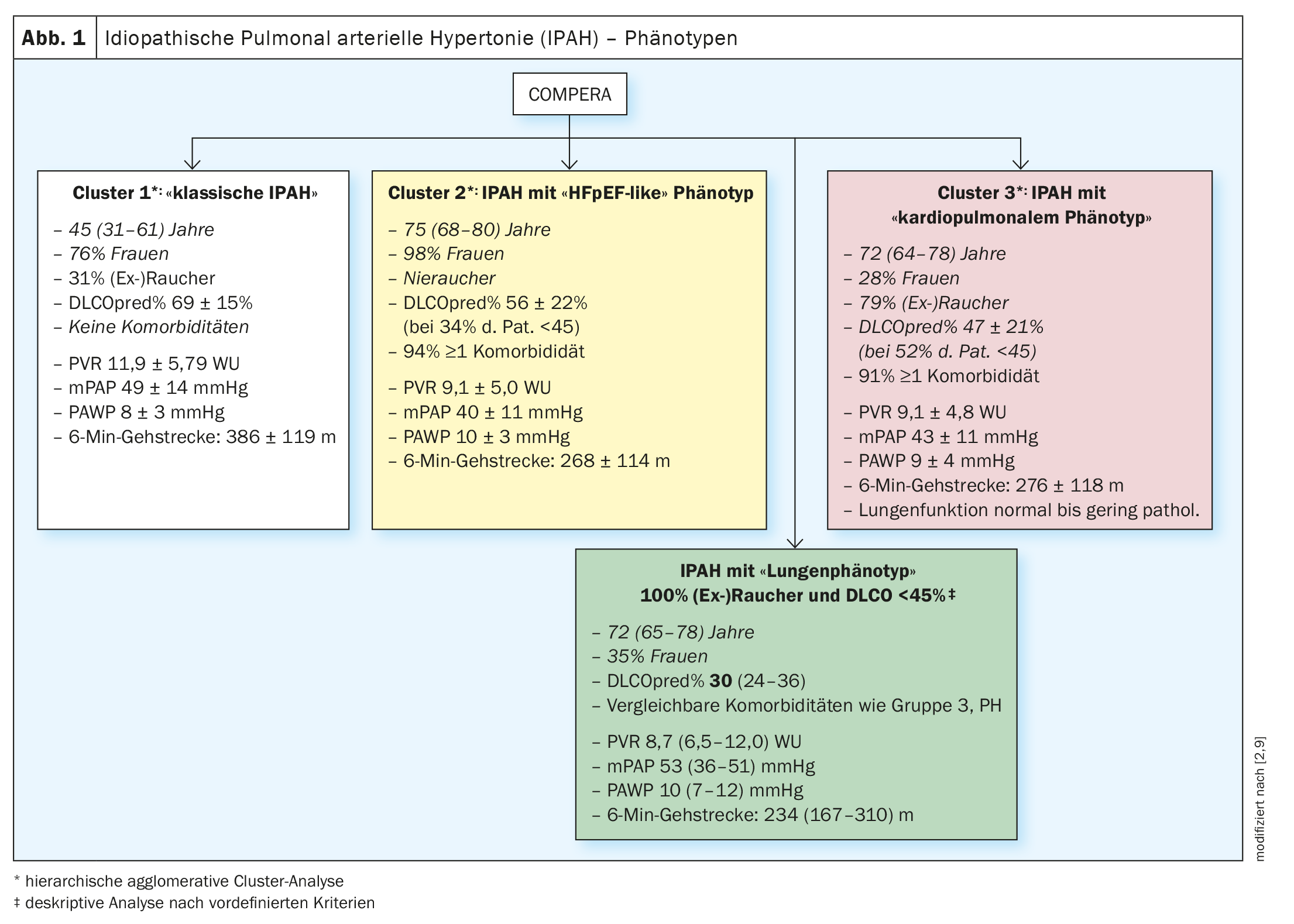
Tutti e tre i cluster erano caratterizzati dalla presenza di una grave PH precapillare (PAWP mediamente inferiore a 11 mmHg in tutti e 3 i gruppi) e da una funzione polmonare (in gran parte) normale, con i pazienti del cluster 3 che presentavano i parametri funzionali polmonari più bassi e l’ipossiemia più pronunciata in confronto. Corretto per l’età, i pazienti del cluster 3 (pazienti con IPAH con fenotipo cardiopolmonare) avevano una prognosi significativamente peggiore rispetto ai pazienti del cluster 1 (pazienti con IPAH senza comorbidità) [2].
Sebbene non tutti i pazienti del cluster 3 nel registro COMPERA avessero una DLCO <45% (53% dei casi) e uno stato positivo (ex)di fumatore (79% dei casi) in questa analisi del cluster, in un altro studio di registro PH una DLCO <45% e uno stato positivo (ex)di fumatore erano necessari per definizione per una diagnosi di IPAH con fenotipo polmonare [9]. Per questo studio, è stato analizzato principalmente il database del registro COMPERA e il registro ASPIRE è servito come validazione indipendente dei risultati [9].
In questo studio, è stato dimostrato che il gruppo di pazienti con IPAH con un “fenotipo polmonare” era molto più simile ai pazienti del gruppo 3-PH (PH associata a una malattia polmonare significativa) in termini di risposta al trattamento e di prognosi rispetto ai pazienti con la cosiddetta IPAH “classica” (IPAH senza fattori di rischio per HFpEF) [9].
L’analisi dei dati di COMPERA ha mostrato, come nell’analisi precedente [2], che i pazienti con IPAH “classica” (senza fattori di rischio per HFpEF) erano prevalentemente più giovani (mediana 45 anni) e soprattutto donne (77%). Circa 1/3 di loro aveva una storia di fumo con una mediana di 14 anni di pacchetto (py), ma una funzione polmonare conservata e una DLCO solo leggermente ridotta (69% mediana). Dal punto di vista emodinamico, questi pazienti erano caratterizzati da una grave ipertensione polmonare precapillare (mPAP 48 mmHg, PVR 10,9 unità di legno (WU)) e da una capacità di esercizio funzionale moderatamente ridotta (distanza di cammino di 6 minuti: 410 m) [9].
In confronto, i pazienti con IPAH con fenotipo polmonare erano più anziani (mediana 72 anni) e più spesso di sesso maschile (65%). Tutti i pazienti con fenotipo polmonare erano fumatori per definizione e avevano una mediana di 40 anni. FVC e FEV1 erano mediani nel range inferiore di normalità o leggermente ridotti e significativamente inferiori rispetto ai pazienti con IPAH classica. La DLCO era significativamente compromessa al 30% mediano e la paO2era di 56 mmHg mediani. Rispetto ai pazienti con IPAH classica, l’emodinamica era meno compromessa (mPAP 43 mmHg, PVR 8,7 WU), ma la capacità di esercizio funzionale (distanza di cammino di 6 minuti) era significativamente più compromessa (234 m). (Fig. 1 – casella verde).
I pazienti con PH del Gruppo 3 in questo studio avevano un’età mediana paragonabile a quella dei pazienti con fenotipo polmonare (71 anni contro 72 anni di media) e una distribuzione di genere paragonabile (63% contro 65% di uomini) [9]. L’81% erano (ex)fumatori con una mediana di 40 py, mentre qui c’erano limitazioni della funzione polmonare di FEV1 e FVC e la DLCO era più ridotta con una mediana del 26%. Al contrario, l’emodinamica era relativamente meno limitata (mPAP 39 mmHg e PVR 7,4 WU mediani) e la capacità di esercizio funzionale (distanza del cammino di 6 minuti) era paragonabile a quella dei pazienti con fenotipo polmonare (238 m vs 234 mediani).
I cambiamenti morfologici della TAC in termini di enfisema o malattia polmonare interstiziale nel gruppo di pazienti ASPIRE sono aumentati in modo significativo dal gruppo di pazienti con IPAH “classica” al gruppo 3 di pazienti con PH (dal 10% a circa il 60%) [9].
Tutti e tre i gruppi di PH hanno mostrato chiare differenze in termini di risposta al trattamento e prognosi. Nei pazienti con IPAH classica rispetto ai pazienti con IPAH con fenotipo polmonare e rispetto ai pazienti con PH del gruppo 3, le probabilità di sopravvivenza a 3 anni erano del 90% contro il 49% contro il 43% e le probabilità di sopravvivenza a 5 anni erano dell’84% contro il 31% contro il 26%. Gli autori hanno suggerito che la causa dell’insufficienza cardiaca destra nei pazienti caratterizzati da IPAH con fenotipo polmonare fosse l’ipertensione polmonare indotta dal fumo di sigaretta e hanno concluso che questi pazienti dovrebbero probabilmente essere classificati più come pazienti con PH di gruppo 3 e che il ruolo dei farmaci specifici per la PH in questi pazienti non è chiaro [9].
Sommario
In sintesi, sembra probabile che i pazienti (ex)fumatori che soddisfano gli attuali criteri diagnostici per l’IPAH, ma che presentano una riduzione della DLCO molto grave che non può essere spiegata altrimenti e/o anche solo lievi anomalie parenchimali alla TAC del torace, si trovino in un gruppo PH diverso (probabilmente il gruppo PH 3) rispetto ai pazienti con IPAH classica (gruppo PH 1).
Presumibilmente, la grave insufficienza cardiaca destra in questi pazienti è causata dal danno all’arteria polmonare dovuto all’inalazione di fumo di sigaretta. È probabile che l’uso di farmaci specifici per la PH sia significativamente meno efficace nei pazienti con la forma più probabile di ipertensione polmonare associata al fumo, rispetto ai pazienti con IPAH classica.
Poiché questi pazienti sono stati esclusi o sottorappresentati nei precedenti studi pivotali sui farmaci specifici per la PH, a causa dei criteri di inclusione ed esclusione, sono urgentemente necessari studi prospettici, randomizzati e controllati con placebo, sia per caratterizzare questi pazienti in modo più preciso, sia per chiarire le opzioni di trattamento e il valore aggiunto dei farmaci specifici per la PH.
Messaggi da portare a casa
- L’attuale gruppo di pazienti con IPAH è più eterogeneo di quanto si pensasse in precedenza.
- I pazienti che soddisfano i criteri per l’IPAH, ma che presentano prove di PH associata al fumo, dovrebbero essere oggetto di maggiore attenzione scientifica in futuro, per chiarire a quale gruppo di PH appartengono questi pazienti e quali opzioni terapeutiche efficaci esistono.
- Sono urgentemente necessari studi di trattamento prospettici, controllati con placebo e randomizzati per chiarire le opzioni terapeutiche per la PH presumibilmente associata al fumo.
Letteratura:
- Lewis RA, Thompson AAR, Billings CG, et al: La malattia polmonare parenchimale lieve e/o la bassa capacità di diffusione hanno un impatto sulla sopravvivenza e sulla risposta al trattamento nei pazienti con diagnosi di ipertensione arteriosa polmonare idiopatica. European Respiratory Journal; doi: 10.1183/13993003.00041-2020.
- Hoeper MM, Pausch C, Grünig E, et al: Fenotipi di ipertensione arteriosa polmonare idiopatica determinati dall’analisi dei cluster del registro COMPERA. Journal of Heart and Lung Transplantation 2020; 39: 1435-1444; doi: 10.1016/j.healun.2020.09.011.
- Trip P, Nossent EJ, De Man FS, et al: Capacità di diffusione gravemente ridotta nell’ipertensione arteriosa polmonare idiopatica: caratteristiche del paziente e risposte al trattamento. European Respiratory Journal; doi: 10.1183/09031936.00184412.
- Chandra S, Shah SJ, Thenappan T, et al: Capacità di diffusione del monossido di carbonio e mortalità nell’ipertensione arteriosa polmonare. Journal of Heart and Lung Transplantation 2010; 29: 181-187; doi: 10.1016/J.HEALUN.2009.07.005.
- Olsson KM, Fuge J, Meyer K, et al: Ancora sull’ipertensione arteriosa polmonare idiopatica con una bassa capacità diffusiva. European Respiratory Journal 2017; 50; doi: 10.1183/13993003.00354-2017.
- Hoeper MM, Apitz C, Grünig E, et al: Terapia mirata dell’ipertensione arteriosa polmonare. DMW 2016; S33-S41.
- Hoeper MM, Apitz C, Grünig E, et al: Terapia mirata dell’ipertensione arteriosa polmonare: raccomandazioni aggiornate dalla Conferenza di consenso di Colonia 2018. Int J Cardiol 2018; 272: 37-45; doi: 10.1016/j.ijcard.2018.08.082.
- Trip P, Nossent EJ, De Man FS, et al: Capacità di diffusione gravemente ridotta nell’ipertensione arteriosa polmonare idiopatica: caratteristiche del paziente e risposte al trattamento. European Respiratory Journal 2013; 42: 1575-1585; doi: 10.1183/09031936.00184412.
- Hoeper MM, Dwivedi K, Pausch C, et al: Fenotipizzazione dell’ipertensione arteriosa polmonare idiopatica: un’analisi del registro. Lancet Respir Med 2022; 10(10): 937-948; doi: 10.1016/S2213-2600(22)00097-2.
COI:
- Halank: onorari per attività di docenza e consulenza da AstraZeneca, Janssen e MSD. Spese di viaggio di Janssen. Il tutto non in relazione all’attuale documento di revisione
- Heberling: onorario per attività di docenza e consulenza e spese di viaggio da Janssen-Cillag e MSD.
- Kolditz: nessuna in relazione a questo lavoro
- Koschel: nessuno in relazione a questo lavoro
InFo PNEUMOLOGIE & ALLERGOLOGIE 2023; 5(3): 6–11