Se uma ou ambas as câmaras do coração forem excessivamente espessas ou dilatadas, o coração já não é tão eficiente. As cardiomiopatias caracterizam-se por uma variedade de causas diferentes de tais alterações no tecido muscular do coração. No entanto, a diferenciação das manifestações clínicas individuais tem grandes implicações para o prognóstico e possíveis regimes de tratamento.
As cardiomiopatias têm muitas faces. Basicamente, podem ser classificados em quatro fenótipos morfológicos com cardiomiopatia hipertrófica (HCM), cardiomiopatia dilatada (DCM), cardiomiopatia arritmogénica (ARVC) e cardiomiopatia restritiva (RCM). No entanto, esta classificação aproximada, baseada na visão, tem muito pouco em comum com as causas reais, como se pode ver pelo grande número de subcategorias. O Prof. Dr. Med. Benjamin Meder, Heidelberg (D), mostrou que a imagiologia por si só não é, portanto, suficiente para detectar a doença. Por conseguinte, as características funcionais devem ser acrescentadas. Isto porque a cardiomiopatia é, em última análise, a descrição de um fenótipo morfológico e funcional do miocárdio que não pode ser explicado por doença arterial coronária ou propriedades de enchimento alteradas devido a hipertensão arterial, viciação valvular ou doença cardíaca congénita.
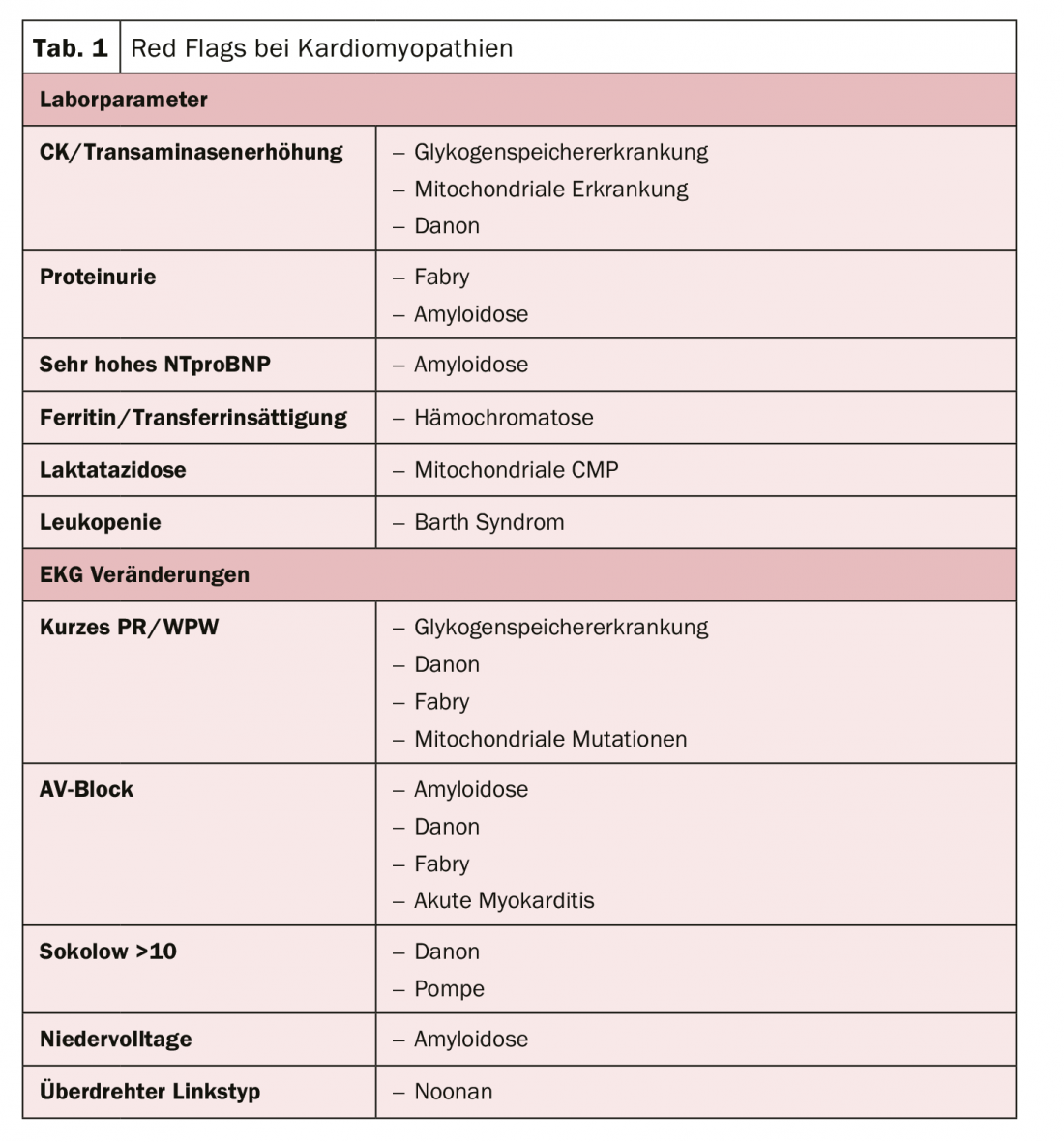
A abordagem estrutural do tratamento optimizado aborda, portanto, através da etiologia com o objectivo de evitar a morte cardíaca súbita – o problema limitador de todas as cardiomiopatias. Existem parâmetros laboratoriais típicos e anomalias no ECG que podem indicar a presença de uma cardiomiopatia (tab. 1). Ser capaz de fazer estas distinções é portanto relevante, uma vez que o prognóstico é essencialmente determinado pela causa, diz o perito. Especialmente os pacientes com amiloidose têm uma sobrevivência muito fraca.
Detectar amiloidose e tratá-la eficazmente
A amiloidose pode ser dividida em três tipos com base no envolvimento de órgãos: Amiloidose AL que afecta os rins, coração e/ou intestinos, amiloidose mt-ATTR que afecta principalmente o coração ou sistema nervoso, e amiloidose wt-ATTR que afecta principalmente o coração. Em princípio, a amiloidose AL pode ser entendida como uma complicação da discrasia de plasmócitos. Portanto, uma terapia hematológica semelhante à do mieloma múltiplo é utilizada com o objectivo de eliminar as cadeias de luz amilóide, (cardio-)tóxicas o mais rapidamente possível. Para além da quimioterapia de alta dose para pacientes em forma, é utilizado principalmente um regime baseado em inibidores de proteasomas. Em pacientes mais jovens, o CyBorD (bortezomib, ciclofosfamida, dexametasona) deve ser favorecido em relação ao BMDex (bortezomib, melphalan, dexametasona) devido à toxicidade das células estaminais do melphalan, a fim de preservar a opção da aférese subsequente das células estaminais e da quimioterapia de dose elevada.
Na amiloidose causada por depósitos de proteína de transthyretin, drogas como a diflunisal e a tafamidis podem estabilizar a proteína mutante. Além disso, as terapias genéticas que reduzem a produção de transthyretin (por exemplo, patisiran, inotersen) podem reduzir os efeitos sobre o sistema nervoso.
Para pacientes com AL cardíaca sintomática e amiloidose ATTR, aplicam-se em princípio as mesmas recomendações terapêuticas gerais que para pacientes com insuficiência cardíaca. No entanto, mesmo doses baixas de β-bloqueadores ou inibidores da ECA podem levar a hipotensão sintomática. Portanto, o tratamento em doentes com amiloidose cardíaca baseia-se principalmente na dosagem correcta de diuréticos.
Envolvimento sindromal de muitos sistemas de órgãos – doença de Fabry
A doença de Fabry é uma doença geneticamente herdada que geralmente se manifesta entre os 20-40 anos de idade. Forma-se uma enzima desdobrada ou não funcional α-Gal A, onde o transporte do retículo endoplasmático para o lisossoma é perturbado. Isto leva a uma acumulação de substratos lisossómicos. Além do coração, os rins, o sistema nervoso central, mas também o sistema nervoso periférico e a pele podem estar envolvidos. Se não for tratada, a insuficiência renal é a causa de morte mais frequente nestes doentes, explicou a Prof. Dra. Ingrid Kindermann, Homburg/Saar (D). Os sintomas cardíacos ocorrem em mais de metade de todos os pacientes de Fabry. As arritmias malignas são as principais responsáveis pela morte cardíaca súbita.
Durante muito tempo, a terapia limitou-se apenas a aliviar os sintomas. Entretanto, existe uma opção de tratamento específico em que a enzima em falta é substituída por uma enzima produzida biotecnologicamente. A alfa-galactosidase A produzida em laboratório é administrada ao doente por infusão e assegura que o material de armazenamento acumulado seja decomposto. No entanto, uma vez que a terapia de substituição enzimática não pode reparar danos de órgãos que já ocorreram, mas apenas atrasar a sua progressão, deve ser utilizada o mais cedo possível. Outra opção é tomar um acompanhante farmacológico oralmente. Este agente pode ser utilizado em doentes com determinadas mutações no gene GLA e actividade residual da enzima α-galactosidase A. Isto porque se liga a formas instáveis de AGAL e estabiliza a enzima de modo a poder decompor as substâncias gordas acumuladas na célula.
Fonte: CardioMedLive 2020
CARDIOVASC 2020; 19(3): 28-29 (publicado 17/9/20, antes da impressão).