Los nódulos tiroideos son un fenómeno bien conocido, los carcinomas medulares de tiroides afortunadamente no. Se trata de una degeneración maligna de las células C de la glándula tiroides, que están situadas parafolicularmente y no pueden almacenar yodo. Dado que segrega calcitonina, ésta puede utilizarse como marcador tumoral.
Los nódulos tiroideos se diagnostican con frecuencia en la consulta, pero afortunadamente el carcinoma medular de tiroides rara vez se diagnostica. Sólo alrededor del 3% de todos los carcinomas tiroideos son carcinomas medulares de tiroides (CMT), que presentan algunas características especiales: Se trata de una degeneración maligna de las células C de la glándula tiroides, que se localizan parafolicularmente y no pueden almacenar yodo; segrega calcitonina (Ctn) y CEA, que se utilizan como marcadores tumorales; una cuarta parte de los casos se dan familiarmente en el contexto de una neoplasia endocrina múltiple de tipo 2 (MEN2). La única opción para una posible curación del CTM es un diagnóstico precoz y una cirugía adecuada. Esto es posible gracias al uso sistemático de la determinación de Ctn en el esclarecimiento del estruma nodoso y en la variante familiar de las pruebas genéticas moleculares del protooncogen RET como parte del cribado familiar. Incluso en aquellos que no se curan, el CTM tiene un pronóstico relativamente favorable con una calidad de vida relativamente buena debido a su lento crecimiento. Una estrategia de seguimiento de “vigilancia activa” adaptada al riesgo es posible en muchos casos. El CTM en la fase de metástasis a distancia progresiva sintomática puede tratarse hoy en día con inhibidores de la tirosina cinasa (ITC) [1,2].
Clínica y diagnóstico del carcinoma medular de tiroides (CMT)
Clínicamente, el CTM medular no difiere significativamente de los demás carcinomas tiroideos: nódulo creciente en la tiroides, molestias inespecíficas en el cuello, desarrollo de inflamación de los ganglios linfáticos cervicales, y en el estadio metastásico avanzado, puede producirse una diarrea pronunciada inducida por el tumor. Hoy en día, el CTM se diagnostica como un hallazgo incidental en la histología de una muestra de cirugía tiroidea o durante el examen preoperatorio como parte del estudio de un nódulo tiroideo. (Fig.1). Los indicadores son un nódulo sospechoso en la ecografía tiroidea (eco pobre, microcalcificaciones, márgenes difusos, extensión más profunda que ancha), unos ganglios linfáticos cervicales sospechosos, una aspiración con aguja fina sospechosa y un nivel de Ctn elevado. Un nivel de Ctn superior a 100 pg/ml es el hallazgo más sensible y específico. Los afectados son los pacientes del 4. y 5ª década de vida. En los pacientes más jóvenes, debe considerarse la variante familiar (MEN2), que en ocasiones puede manifestarse a través de afecciones concomitantes como el feocromocitoma y el hiperparatiroidismo primario. El MTC poco frecuente también debe considerarse en el contexto de la aclaración de una elevación del ACE.
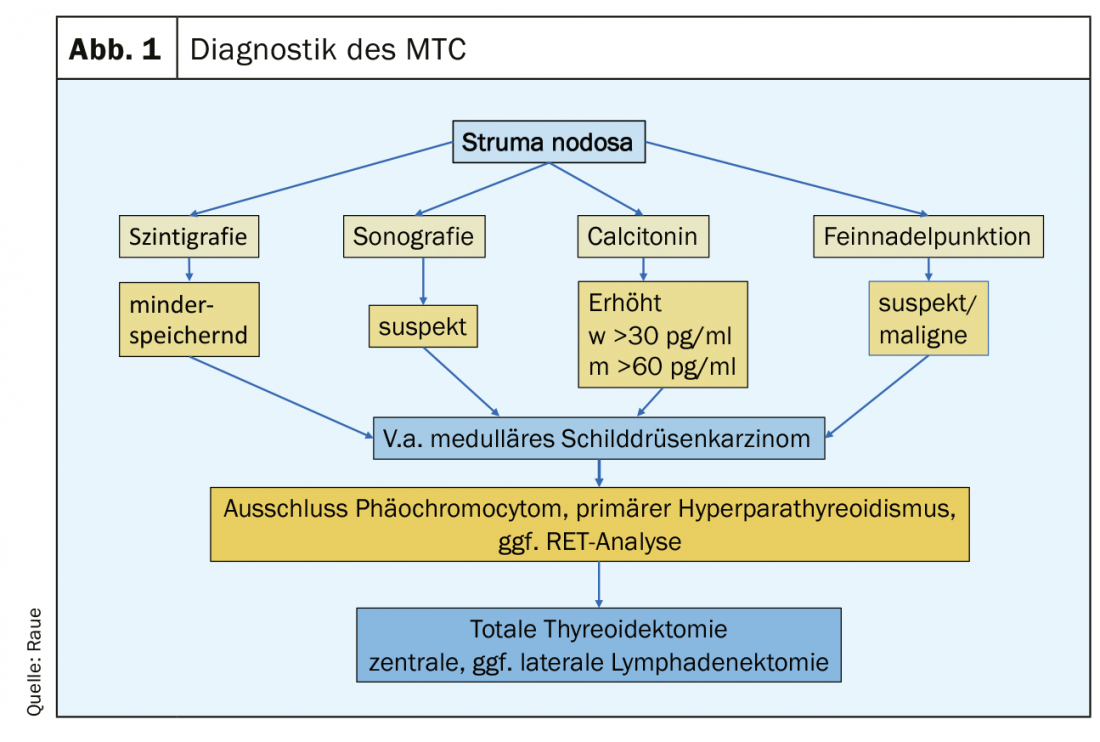
Cribado de calcitonina en el esclarecimiento del estruma nodoso
La Ctn se correlaciona con la masa tumoral y es un marcador tumoral sensible y específico para la detección precoz y el seguimiento del CMT. La determinación rutinaria de la Ctn en la exploración del estruma nodoso hace posible un diagnóstico precoz [3]. Los niveles de Ctn ligeramente elevados se encuentran en la hiperplasia de células C, que puede ser un precursor del CMT hereditario, pero también se observa como un fenómeno concomitante “benigno” en otras enfermedades tiroideas [4]. Con el desarrollo de nuevos ensayos de Ctn de medición sensible y específica totalmente automatizados (inmunoensayos quimioluminiscentes con una sensibilidad de 0,5 pg/ml y valores de referencia superiores específicos para cada sexo) [5,6], los niveles basales de Ctn han adquirido mayor importancia para el cribado en el estruma nodoso.
En niveles preoperatorios de Ctn superiores a 100 pg/ml, el CMT está presente en casi el 100% de los casos, mientras que en niveles de Ctn entre 10-20 pg/ml, la tasa de hallazgo de CMT es inferior al 5% [7]. Teniendo en cuenta la sensibilidad y la especificidad, los valores de corte específicos por sexo de la Ctn basal para la recomendación de cirugía de una sospecha de CMT resultan en aprox. 30 pg/ml para las mujeres y aprox. 60 pg/ml para los hombres. El intervalo gris de Ctn para las mujeres es de 20-30 pg/ml, para los hombres de 30-60 pg/ml, en el que se pasaron por alto entre un 6 y un 13% de CMT pequeños [8]. El aumento de los niveles de Ctn es más indicativo de un MTC, en cuyo caso debería recomendarse la cirugía. Las medidas quirúrgicas pueden curar el CMT en casi el 100% hasta un nivel de Ctn de 100 pg/ml [9].
Mutaciones de la línea germinal del protooncogén RET en MEN2
En aproximadamente el 25% de los pacientes con CMT, puede detectarse una mutación de la línea germinal en el protooncogén RET como indicio del síndrome MEN2. Clínicamente, la MEN2A con el posible desarrollo de un feocromocitoma e hiperparatiroidismo se distingue de la MEN2B con un hábito típico, gangloneuromatosis de las mucosas y el posible desarrollo de un feocromocitoma. No todas las familias pueden ser captadas por un historial familiar, ya que a menudo las familias son pequeñas o se trata de un hMTC de manifestación tardía y baja penetrancia. En un pequeño porcentaje, se observa una mutación “de novo” en el paciente, los demás miembros de la familia no están afectados. En una gran serie, se encontró una mutación de línea germinal en el protooncogén RET en el 12% de los CTM aparentemente esporádicos. Por lo tanto, deben organizarse pruebas genéticas moleculares para detectar mutaciones en la línea germinal del gen RET en todos los pacientes con un CMT.
En la forma hereditaria del CMT, las mutaciones causantes se caracterizan en casi todas las familias, en su mayoría mutaciones puntuales en 8 exones diferentes del protooncogén RET. La detección genética molecular de estas mutaciones, junto con la determinación de la Ctn en las familias de los niños y adolescentes afectados, permite una tiroidectomía precoz con curación del CMT. El momento óptimo de la tiroidectomía en portadores de una mutación RET se recomienda hoy en día en función de la clasificación del riesgo de la mutación RET específica en moderado, alto y máximo riesgo de desarrollo precoz de CMT (penetrancia precoz o tardía) (Tab. 1) [1]. El nivel de Ctn define individualmente la tiroidectomía profiláctica que debe planificarse en una fase temprana, de modo que en el caso óptimo no sea necesaria una linfadenectomía adicional.

Hasta el 50% de los pacientes con MEN2 desarrollan feocromocitomas a lo largo de su vida, en función del genotipo, sobre todo tras la manifestación del MTC. Los feocromocitomas pueden ser multifocales y bilaterales [10]. Los pacientes con mutaciones RET-634 y -918 se ven particularmente afectados, siendo menos frecuente con mutaciones en los exones 13-15. Hasta un 10% de los pacientes con MEN2 pueden desarrollar hiperparatiroidismo primario en función de su genotipo; los pacientes con una mutación RET-918 no se ven afectados. Otras manifestaciones extratiroideas, como la ganglioneuromatosis en MEN2B o la liquen amiloidosis interescapular, también dependen del genotipo.
En el tejido tumoral del CTM esporádico se detecta con frecuencia una mutación somática en el gen RET, principalmente RET-M918T, curiosamente con menor frecuencia en los tumores más pequeños, con mayor frecuencia en los más grandes y con mayor frecuencia en las filias de los ganglios linfáticos y en las metástasis a distancia, lo que sugiere que esta mutación no es el acontecimiento primario en la tumorigénesis. En el tejido tumoral en el que no se encuentra una mutación RET, la detección de una mutación RAS suele tener éxito. La detección de la mutación somática RET tiene importancia pronóstica y es un requisito previo para la terapia con los inhibidores selectivos de la tirosina quinasa RET (LOXO-292, BLU-667) que probablemente pronto estarán disponibles [11].
Funcionamiento del MTC
La terapia primaria para el MTC es quirúrgica. La tiroidectomía total es el procedimiento mínimo, complementado con la disección de los ganglios linfáticos (Ctn) central y, si es necesario, lateral unilateral/bilateral, en función del Ctn medido preoperatoriamente y del estadio tumoral. En el caso de un CTM descubierto por casualidad en la histología, el nivel de Ctn postoperatorio debe decidir el procedimiento ulterior; si el Ctn no es mensurablemente bajo, no es necesaria ninguna cirugía ulterior; si el Ctn es elevado, en función del estadio tumoral, debe realizarse una cirugía completa (TX total y disección central y lateral del LK). La terapia quirúrgica curativa sólo es posible si no se han descrito metástasis a distancia, ni infiltración en los tejidos blandos en la histología primaria y si se detectan menos de 5-10 LK cervicales afectados en la histología previa o menos de 3 compartimentos afectados tras la linfadenectomía sistemática [12]. Si se da esta situación favorable, debe intentarse una terapia quirúrgica curativa. En el caso de un CMT hereditario, una tiroidectomía total es apropiada en todos los casos, ya que en principio un CMT puede desarrollarse a partir de cada célula C. Del CTM diagnosticado a través de un nódulo tiroideo palpable, el 70% ya tiene metástasis en los ganglios linfáticos cervicales y el 13% tiene metástasis a distancia en hígado, pulmón o hueso [13]. La supervivencia a 10 años es aproximadamente del 61-81% [14], dependiendo del estadio tumoral en el momento del diagnóstico, la edad, el sexo y los niveles de Ctn pre y postoperatorios. En la última década, debido a un diagnóstico cada vez más precoz, a estadios tumorales más favorables y a la mejora de las estrategias quirúrgicas, la supervivencia ha aumentado en general, pero también en los estadios tumorales más elevados [14,15].
En los pacientes con MEN2 detectados por cribado familiar, el momento de la “tiroidectomía profiláctica” depende de la mutación RET y del nivel de Ctn (cf. Tab. 1). El diagnóstico precoz y la cirugía completa aumentaron la supervivencia a 5 años específica de la enfermedad de todos los CTM hasta el 89% [14].
Cuidados posteriores al MTC
En el postoperatorio, debe disponerse de los siguientes resultados para planificar un seguimiento estructurado: la histología, en su caso inmunohistología Ctn del tumor, la clasificación según el esquema pTNM, el resultado del análisis RET para la clasificación en la variante hereditaria o esporádica y el nivel de Ctn postoperatorio. Se pueden definir tres grupos de pacientes de riesgo en términos de respuesta a la terapia primaria y pronóstico: excelente (Ctn no mensurablemente bajo), bioquímicamente incompleto (Ctn mensurable, pero sin tejido tumoral detectable por imagen) y estructuralmente incompleto (Ctn significativamente elevado, evidencia de ganglios linfáticos metastásicos o metástasis a distancia). (Fig. 2). En el curso posterior, se calcula el tiempo de duplicación del marcador tumoral (Ctn/CEA) y se realizan pruebas de imagen para detectar un posible crecimiento tumoral, seguido del ajuste del grupo de riesgo [16,17]. Los intervalos de seguimiento se adaptan al riesgo y se realizan cada 3 meses hasta una vez al año, dependiendo del tamaño y la localización del tumor residual/metástasis y del grado de progresión tumoral [18]. Dependiendo de la localización de las metástasis sospechosas, los procedimientos de diagnóstico por imagen incluyen ecografía de cuello y abdomen, tomografía computerizada con medio de contraste, resonancia magnética, gammagrafía ósea y, posiblemente, FDG-PET o F-DOPA-PET (Fig. 1).
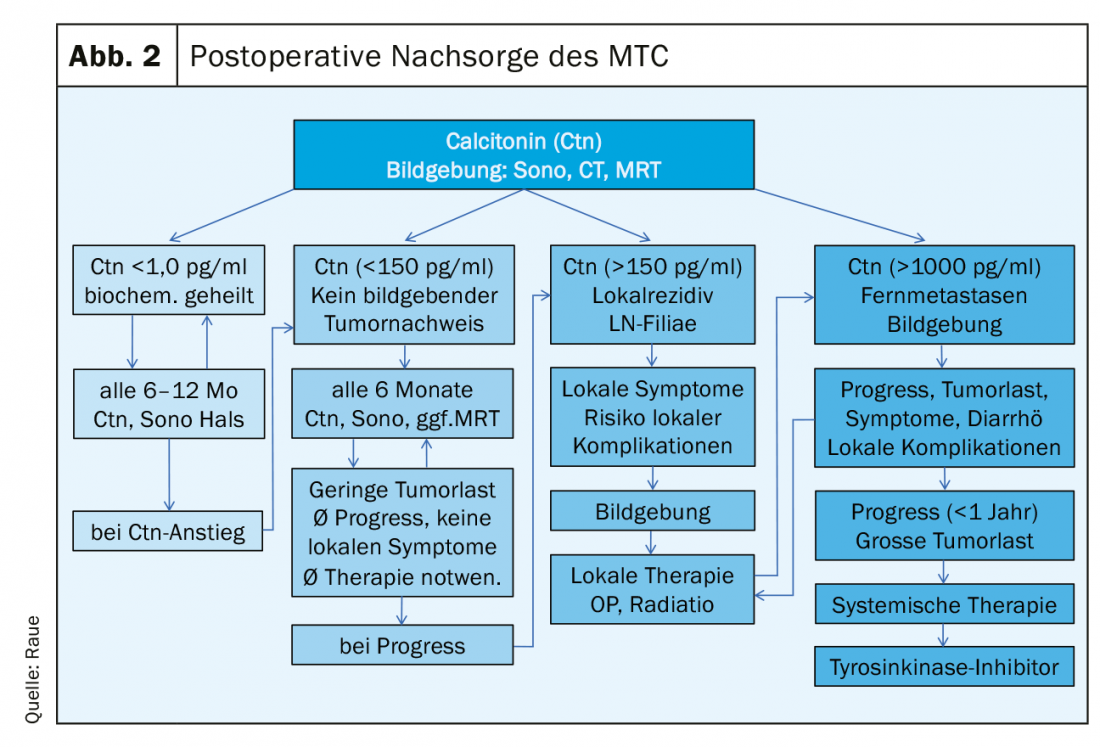
Grupos de riesgo en el seguimiento
En el caso de una respuesta excelente, el nivel de Ctn no es mensurablemente bajo: entonces se puede suponer que el paciente está bioquímicamente curado, siempre que la histología sea correcta (inmunohistología Ctn obligatoria). En los 2 primeros años, son suficientes los exámenes de seguimiento semestrales con ecografía del cuello, determinación de Ctn y CEA, comprobación del tratamiento sustitutivo con tiroxina (objetivo: TSH en el rango normal), y si es necesario, en caso de hipoparatiroidismo postoperatorio, la administración de calcio y calcitriol (objetivo: calcio sérico en el rango límite inferior 2,0-2,3 mmol/l) [19]. A partir de entonces, si no se ha producido ningún aumento de Ctn, es posible pasar a inspecciones anuales. El pronóstico es excelente y no difiere del de la población normal.
En caso de respuesta bioquímicamente incompleta, el nivel de Ctn suele ser persistentemente de leve a moderadamente elevado (normalmente por debajo de 1000 pg/ml). El tejido tumoral restante debe suponerse. Si la operación anterior no fue adecuada, se realiza una “puesta en escena” seguida de una operación complementaria adecuada. Sin embargo, si se superan estos límites (véase la sección Funcionamiento del MTC), ya no es posible un enfoque curativo, por lo que todas las medidas terapéuticas posteriores deben sopesarse de forma crítica, teniendo en cuenta el riesgo de morbilidad y la calidad de vida, que suele ser buena. A menudo, a pesar de una búsqueda intensiva, no se encuentra un correlato tumoral definido con una Ctn moderadamente elevada de hasta 150 pg/ml. La progresión de la enfermedad tumoral en el curso posterior puede ser muy diferente y puede estimarse relativamente bien sobre la base del tiempo de duplicación de Ctn y CEA [20]. Requisito previo para una buena declaración son al menos 4 valores Ctn a lo largo de 2 años. Los pacientes con tiempos de duplicación de marcadores tumorales inferiores a 24 meses también presentaron progresión morfológica por imagen en un 94%. Cuando los tiempos de duplicación de los marcadores tumorales se prolongaron durante 24 meses, en el 86% de los pacientes no se detectó crecimiento tumoral [21]. En el curso posterior, suelen bastar comprobaciones semestrales de los progresos. La frecuencia del diagnóstico por imagen puede planificarse en función de los hallazgos primarios, el crecimiento durante la progresión (criterios RECIST) y el tiempo de duplicación del marcador tumoral. Un diagnóstico de máxima aproximación sin consecuencias terapéuticas no es útil, por ejemplo, la ecografía cada 6 meses, la tomografía computerizada/IRM cada 12 meses suelen ser suficientes.
En caso de respuesta estructuralmente incompleta, el nivel de Ctn es significativamente elevado (normalmente más de 1000 pg/ml): En este caso, debe suponerse la existencia de tejido tumoral local infiltrante, metástasis en los ganglios linfáticos cervicales o metástasis a distancia principalmente en pulmón, hígado y/o hueso. Ya no es posible un enfoque curativo, las medidas paliativas están en primer plano [22].
Las reoperaciones de una recidiva locorregional bajo un enfoque paliativo son especialmente útiles en casos de recidiva local progresiva o de filiaciones ganglionares dolorosas en la región central del cuello (proximidad/infiltración de la tráquea o el esófago) para reducir las complicaciones locales. No tiene sentido someter a cirugía inmediata cada metástasis cervical recién detectada, ya que esto conduce a numerosas operaciones que no logran su objetivo. No hay pruebas de que estas operaciones tengan un efecto positivo en la evolución general, a menudo se asocian a efectos secundarios como paresia recurrente, hipoparatiroidismo y parálisis muscular del brazo. La radioterapia es útil para las recidivas locales o mediastínicas progresivas inoperables. Dado que la cirugía es más difícil después de la radioterapia, siempre debe examinarse primero la operabilidad desde un punto de vista paliativo.
Las metástasis hepáticas son frecuentes, suelen causar pocos síntomas y rara vez son una indicación para la cirugía. Las metástasis hepáticas significativamente progresivas y dolorosas deben tratarse. En la mayoría de los casos, la metástasis es múltiple y difusa. La terapia local mediante (quimio)embolización o radioterapia interna selectiva (SIRT) tiene una eficacia limitada. La terapia sistémica con inhibidores de la tirosina quinasa es útil en casos de progresión rápida.
Las metástasis óseas rara vez son osteolíticas, propensas a fracturas o dolorosas. La radioterapia externa es adecuada para las metástasis óseas dolorosas, y la radioterapia externa o la intervención quirúrgica para el riesgo de fractura. El tratamiento con bifosfonato/denosumab puede utilizarse adicionalmente de forma individualizada, sobre todo en casos de dolor o riesgo de fractura, especialmente en metástasis osteolíticas [23].
La quimioterapia sólo es ligeramente eficaz en el CTM metastásico. Los inhibidores de la tirosina cinasa se han probado en ensayos para el CMT metastásico avanzado durante unos 10 años. Desde mayo de 2012, el inhibidor de la tirosina quinasa vandetanib está disponible en Suiza para el tratamiento del CMT agresivo y sintomático; en algunos países, entre ellos Alemania, el cabozantinib también ha sido aprobado para el CMT progresivo y está indicado en este país para el tratamiento del carcinoma de células renales avanzado desde 2017. Dado que se carece claramente de pruebas de beneficio en la enfermedad en fase inicial, la indicación de terapia con ITK se considera actualmente en casos de alta carga tumoral y enfermedad progresiva (criterios RECIST, tiempo de duplicación del marcador tumoral corto), así como marcadamente sintomática, cuando se han agotado las medidas de terapia local. Todavía no se ha demostrado la prolongación de la supervivencia con ningún TKI en el MTC metastásico [11].
A pesar de las metástasis a distancia, muchos pacientes tienen una buena calidad de vida durante años porque el crecimiento del tumor suele ser muy lento. Una estrategia de “vigilancia activa” es posible. El tratamiento del CTM metastásico está marcadamente orientado a los síntomas, lo que incluye un tratamiento antidiarreico constante en el estadio tumoral avanzado con loperamida y/o tinctura opii, que a menudo se descuida o se dosifica con demasiada precaución en detrimento de la calidad de vida.
Cuidados de seguimiento para MEN2
Además del seguimiento de la MTC, se realiza un diagnóstico anual con respecto al feocromocitoma a partir del 11º año de vida para las mutaciones RET de alto y máximo riesgo y a partir del 16º año de vida para las mutaciones RET de riesgo moderado mediante la determinación de metanefrinas y catecolaminas y, si es necesario, imágenes de resonancia magnética. En cuanto al hiperparatiroidismo primario, se realiza un chequeo anual con determinación de calcio sérico y parathormona de forma análoga con respecto a la edad en los grupos de riesgo de mutación correspondientes (tab. 1).
Mensajes para llevarse a casa
- El marcador tumoral del carcinoma medular de tiroides (CMT) es la calcitonina.
- El cribado de calcitonina en el estruma nodoso permite el diagnóstico precoz del CMT.
- Todos los pacientes con CMT deben someterse a un análisis genético molecular del protooncogén RET en el momento del diagnóstico.
- Por regla general, el tratamiento de elección es la tiroidectomía total con linfadenectomía central.
- En el seguimiento, se diferencia entre bioquímicamente curada (Ctn no mensurablemente baja), bioquímicamente incompleta (Ctn aumentada sin evidencia tumoral en imagen) y evidencia tumoral estructural (metástasis en imagen).
- En muchos casos con metástasis, es posible la “vigilancia activa”; en caso de progresión y síntomas, se aplica lo siguiente: local (cirugía paliativa, radiación) antes que sistémica (inhibidores de la tirosina quinasa).
Literatura:
- Wells SA Jr, et al: Directrices revisadas de la Asociación Americana de Tiroides para el tratamiento del carcinoma medular de tiroides. Tiroides 2015; 25(6): 567-610.
- Ceolin L, et al: El carcinoma medular de tiroides más allá de la cirugía: avances, retos y perspectivas. Endocr Relat Cancer 2019; 26(9): R499-R518.
- Raue F, Frank-Raue K: Cribado de la calcitonina en el bocio nodular: límites superiores. Dtsch Arztebl Int 2018; 115(13): 221.
- Costante G, et al.: Determinación de los niveles de calcitonina en la enfermedad de células C: interés clínico y escollos potenciales. Nat Clin Pract Endocrinol Metab 2009; 5(1): 35-44.
- Kratzsch J, Petzold A, Raue F, et al.: Calcitonina y procalcitonina basales y estimuladas mediante diversos ensayos en pacientes con y sin cáncer medular de tiroides. Clin Chem 2011; 57(3): 467-474.
- Kahaly GJ, Algeciras-Schimnich A, Davis TE, et al: United States and European Multicenter Prospective Study for the Analytical Performance and Clinical Validation of a Novel Sensitive Fully Automated Immunoassay for Calcitonin. Clin Chem 2017; 63(9): 1489-1496.
- Mian C, Perrino M, Colombo C, et al: Perfeccionamiento de la prueba del calcio para el diagnóstico del cáncer medular de tiroides: valores límite, procedimientos y seguridad. J Clin Endocrinol Metab 2014; 99(5): 1656-1664.
- Frank-Raue K, Schott M, Raue F. En nombre de la Sección de Tiroides de la DGE. [Recommendation for Calcitonin Screening in Nodular Goiter]. Dtsch Med Wochenschr 2018; 143(15): 1065-1069.
- Machens A, Dralle H. Estratificación del riesgo basada en biomarcadores para el cáncer medular de tiroides no tratado previamente. J Clin Endocrinol Metab 2010; 95(6): 2655-2663.
- Mucha L, Leidig-Bruckner G, Frank-Raue K, et al: Feocromocitoma en la neoplasia endocrina múltiple tipo 2: penetrancia específica del codón RET y cambios en el tratamiento durante las últimas cuatro décadas. Clin Endocrinol (Oxf) 2017; 87(4): 320-326.
- Cabanillas ME, Ryder M, Jiménez C: Terapia dirigida para el cáncer de tiroides avanzado: los inhibidores de la cinasa y más allá. Endocr Rev 2019; 40(6): 1573-1604.
- Machens A, Gimm O, Ukkat J, et al: Mejora de la predicción de la normalización de la calcitonina en pacientes con carcinoma medular de tiroides mediante el análisis cuantitativo de los ganglios linfáticos. Cáncer 2000; 88(8): 1909-1915.
- Roman S, Lin R, Sosa JA: Pronóstico del carcinoma medular de tiroides: predictores demográficos, clínicos y patológicos de la supervivencia en 1252 casos. Cáncer 2006; 107(9): 2134-2142.
- Randle RW, Balentine CJ, Leverson GE et al: Tendencias en la presentación, el tratamiento y la supervivencia de los pacientes con cáncer medular de tiroides en los últimos 30 años. Cirugía 2017; 161(1): 137-146.
- Opsahl EM, Akslen LA, Schlichting E, et al: Trends in Diagnostics, Surgical Treatment, and Prognostic Factors for Outcomes in Medullary Thyroid Carcinoma in Norway: A Nationwide Population-Based Study. Eur Thyroid J 2019; 8(1): 31-40.
- Lindsey SC, Ganly I, Palmer F, et al: La respuesta a la terapia inicial predice los resultados clínicos en el cáncer medular de tiroides. Tiroides 2015; 25(2): 242-249.
- Yang JH, Lindsey SC, Camacho CP, et al: La integración de una medición postoperatoria de la calcitonina en un sistema de estadificación anatómica mejora la estratificación inicial del riesgo en el cáncer medular de tiroides. Clin Endocrinol (Oxf) 2015; 83(6): 938-942.
- Raue F, Frank-Raue K: Seguimiento a largo plazo en el carcinoma medular de tiroides. Resultados recientes Cancer Res 2015; 204: 207-225.
- Leidig-Bruckner G, Bruckner T, Raue F, et al: Seguimiento a largo plazo y tratamiento del hipoparatiroidismo permanente postoperatorio en pacientes con carcinoma medular de tiroides: diferencias en la enfermedad completa y parcial. Horm Metab Res 2016.
- www.thyroid.org/professionals/calculators/thyroid-cancer-carcinoma
- Laure Giraudet A, Al Ghulzan A, Auperin A, et al.: Progresión del carcinoma medular de tiroides: evaluación con calcitonina y tiempos de duplicación del antígeno carcinoembrionario. Eur J Endocrinol 2008; 158(2): 239-246.
- Hadoux J, Pacini F, Tuttle RM, et al: Tratamiento del cáncer medular de tiroides avanzado. Lancet Diabetes Endocrinol 2016; 4(1): 64-71.
- Vogel T, Wendler J, Frank-Raue K, et al: Metástasis óseas en el carcinoma medular de tiroides: alta morbilidad y mal pronóstico asociados a la morfología osteolítica. J Clin Endocrinol Metab 2020; 105(6).
InFo ONCOLOGÍA Y HEMATOLOGÍA 2020; 8(3): 6-10