Este artículo ofrece una visión general de las distintas formas de dermatosis bullosas autoinmunes y un resumen de las recomendaciones terapéuticas y opciones de tratamiento actualmente válidas.
Las enfermedades autoinmunes ampollosas suelen ser enfermedades graves de la piel, y a menudo también de las mucosas. Se caracterizan por la aparición de autoanticuerpos IgG o IgA contra proteínas estructurales de la piel. Estas proteínas estructurales son de gran importancia para la adhesión celular de los queratinocitos (enfermedades del pénfigo) o para la adhesión de la epidermis con la dermis (enfermedades del penfigoide, epidermólisis bullosa adquirida, dermatitis herpetiforme).
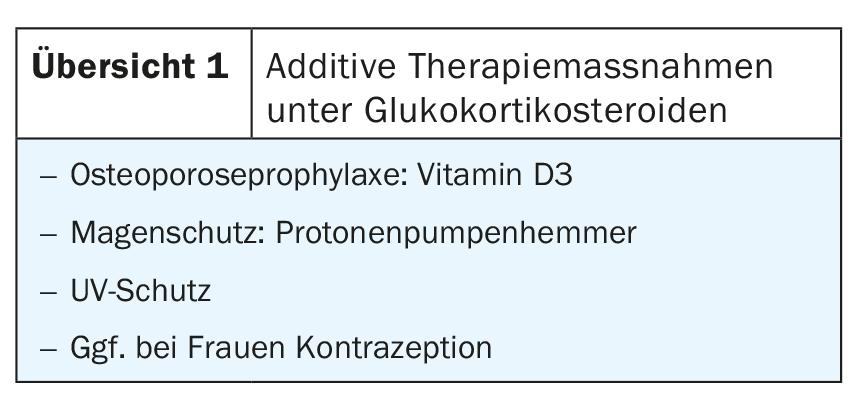
Así, la formación de ampollas se produce intraepidérmicamente en las enfermedades del pénfigo y subepidérmicamente en las demás enfermedades autoinmunes bullosas. La gran heterogeneidad clínica y los diferentes cursos también representan un reto terapéutico para los dermatólogos. Los glucocorticosteroides locales y orales son el tratamiento de primera línea, excepto para la dermatitis herpetiforme. Las medidas terapéuticas aditivas son necesarias bajo terapia sistémica con corticosteroides (resumen 1). Para ahorrar corticosteroides, éstos se combinan con otros inmunosupresores en el curso.
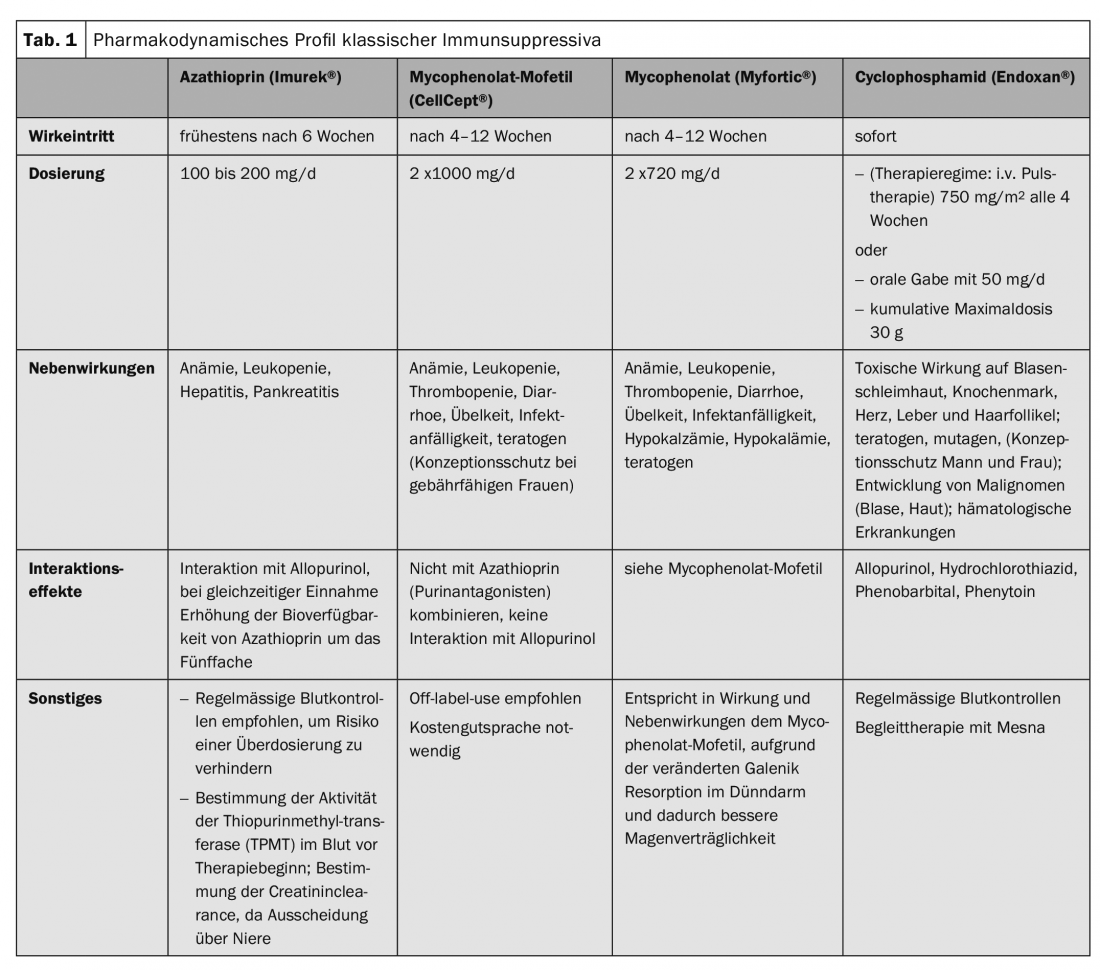
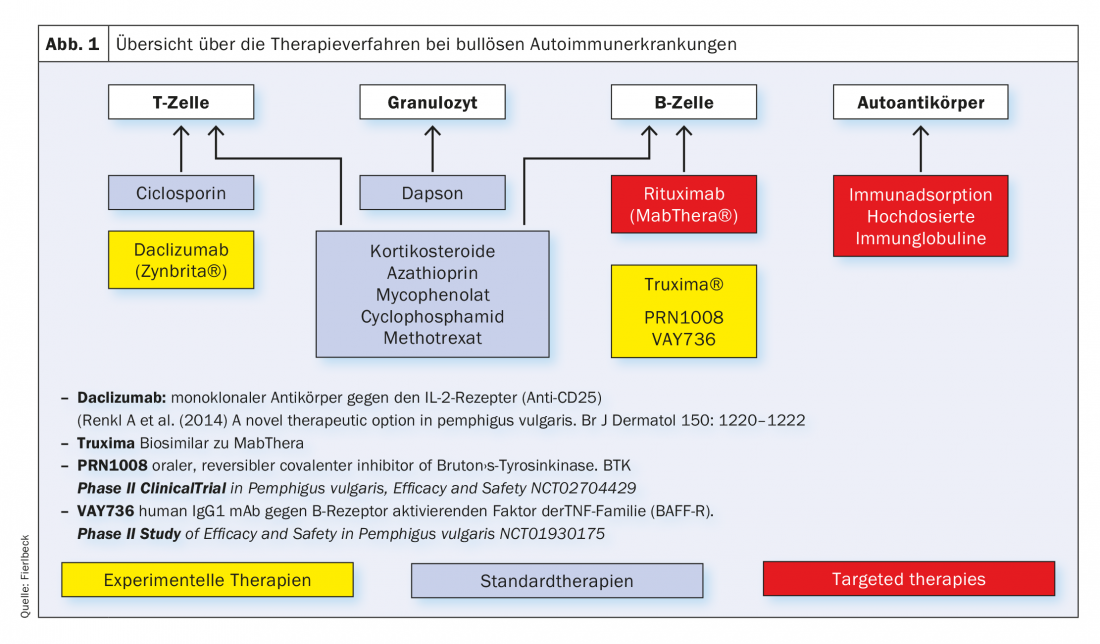
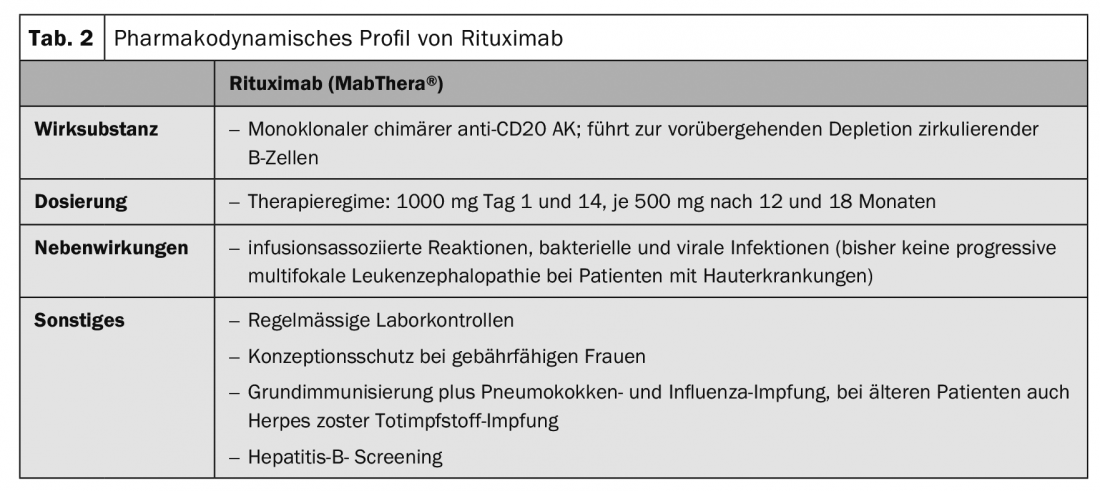
En los últimos años, los fármacos inmunosupresores clásicos han sido (Tab. 1), que influyen en los procesos metabólicos de las células T y B a nivel celular, complementados con terapias dirigidas. (Fig.1). Esencialmente, se trata de rituximab (Tab.2), Proceso de inmunoadsorción (Tab. 3) y el uso de altas dosis de inmunoglobulinas. (Tab.4). El rituximab es un anticuerpo quimérico anti-CD20 para el agotamiento de las células B periféricas que forman autoanticuerpos (Tab.2).
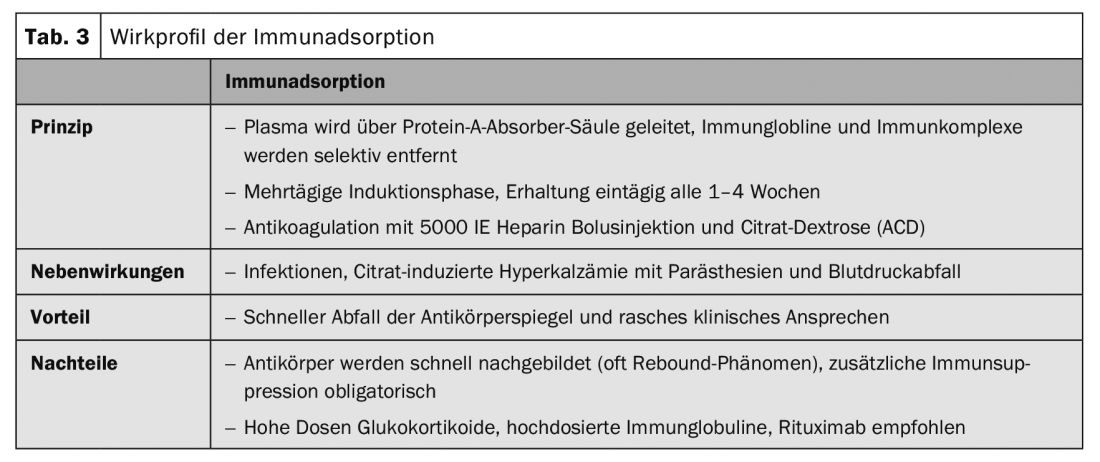
En los procedimientos de inmunoadsorción, los autoanticuerpos y los complejos inmunes se eliminan selectivamente del plasma mediante absorbentes de alta afinidad (tab. 3).
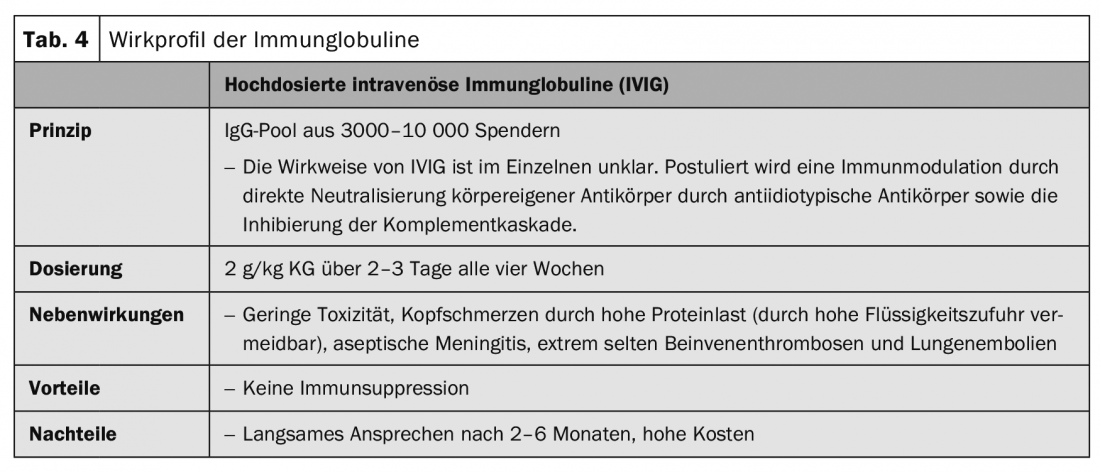
Las altas dosis de inmunoglobulinas intravenosas están destinadas a neutralizar los mediadores inflamatorios y los autoanticuerpos (tab. 4).
Durante la terapia con inmunosupresores, existe un mayor riesgo de reactivación de infecciones latentes, por lo que infecciones como el VIH, la hepatitis B, la tuberculosis y las infecciones bacterianas crónicas deben excluirse antes de iniciar la terapia. Durante la terapia pueden producirse infecciones oportunistas por cándida e infecciones por herpes (herpes simple, varicela zoster). Se ha descrito un mayor riesgo de trastornos linfoproliferativos y tumores cutáneos durante la terapia a largo plazo con inmunosupresores. Debido al aumento de la fotocarcinogénesis, es necesario protegerse de los rayos UV y someterse a revisiones cutáneas periódicas. Antes de iniciar la terapia inmunosupresora, compruebe el estado de vacunación del paciente. Las vacunaciones con vacunas vivas durante la terapia inmunosupresora están contraindicadas, y las vacunaciones con vacunas atenuadas pueden tener un éxito reducido. En pacientes mayores de 50 años, está indicada la vacunación contra el herpes zóster con la vacuna contra la subunidad-tot del herpes zóster [1].
Enfermedades del pénfigo
El pénfigo vulgar, el pénfigo foliáceo y el pénfigo paraneoplásico se subsumen en este término general. Los glucocorticosteroides sistémicos son el centro de la terapia. Dependiendo de la gravedad de la enfermedad, las dosis iniciales corresponden a 1-2 mg/kg equivalentes de prednisolona al día. La terapia de consolidación depende de la actividad de la enfermedad. Si no aparecen nuevas ampollas en 8 días, la dosis de corticoides se reduce un 25% cada 2-4 semanas. A partir de 30 mg equivalentes de prednisolona al día, la reducción posterior es aún más lenta. El objetivo es alcanzar el umbral de Cushing (7,5 mg de prednisolona). Para ahorrar glucocorticosteroides, éstos se combinan con otros inmunosupresores (tab. 1) . La dosis del inmunosupresor permanece inalterada -siempre que no haya efectos secundarios- durante toda la terapia.
El inmunosupresor adyuvante más común es la azatioprina a una dosis de 100-150 mg/día. En caso de actividad enzimática reducida de la tiopurina metiltransferasa o de administración simultánea de alopurinol, debe reducirse la dosis. Deben tomarse muestras de sangre antes y durante la terapia (tab. 1).
Si aparecen efectos secundarios con la azatioprina, se utiliza el micofenolato mofetilo o, en caso de efectos secundarios gastrointestinales, el ácido micofenólico . Ambos fármacos tienen un efecto ahorrador de esteroides y conducen a una remisión más rápida (tab. 1).
En cursos muy refractarios, la ciclofosfamida puede combinarse con corticosteroides. La ciclofosfamida es una sustancia alquilante, es decir, que entrecruza el ADN, con efectos irreversibles sobre la reserva ovárica y la espermatogénesis, por lo que resulta obsoleta en pacientes jóvenes. Además, pueden producirse una serie de efectos secundarios tóxicos y el desarrollo de neoplasias malignas (Tab. 1). Por ello, la aprobación en la UE se restringió a las enfermedades autoinmunes potencialmente mortales en 2012.
Los ensayos clínicos prospectivos, multicéntricos y aleatorizados con y sin corticosteroides han demostrado la eficacia del rituximab (tab. 2) con remisiones de hasta el 80% tras 24 meses. Los títulos de autoanticuerpos (anti-desmogleína I y III) descendieron lentamente en las primeras semanas tras la infusión y alcanzaron los valores más bajos al cabo de unos 180 días. Se observó una respuesta clínica al cabo de 2-3 meses. Basándose en los estudios, el rituximab fue aprobado para el tratamiento inicial del pénfigo grave y moderado en EE.UU. en junio de 2018 [2,3]. Se espera su aprobación en Europa en los próximos años.
Los resultados del primer estudio sobre inmunoadsorción ( IA) (tab. 3 ) en el pénfigo se publicaron en 2007 [4]. Mientras tanto, se han publicado más de 100 pacientes con diferentes protocolos terapéuticos. En contraste con el rituximab, todas las series de casos mostraron una rápida disminución de los autoanticuerpos y una rápida mejoría clínica, pero también una elevada tasa de recaída al cabo de poco tiempo. La combinación de IA con rituximab dio lugar a remisiones rápidas y duraderas [5].
Las series de casos y los informes de casos individuales han informado de una buena respuesta tras varias series terapéuticas de inmunoglobulinas intravenosas (IGIV) a dosis altas (tab. 4) en combinación con glucocorticosteroides en el pénfigo vulgar y el pénfigo paraneoplásico como terapia de segunda o tercera línea tras el fracaso de la terapia inmunosupresora combinada. Sólo después de varios meses de terapia hubo respuesta.
Penfigoide bulloso
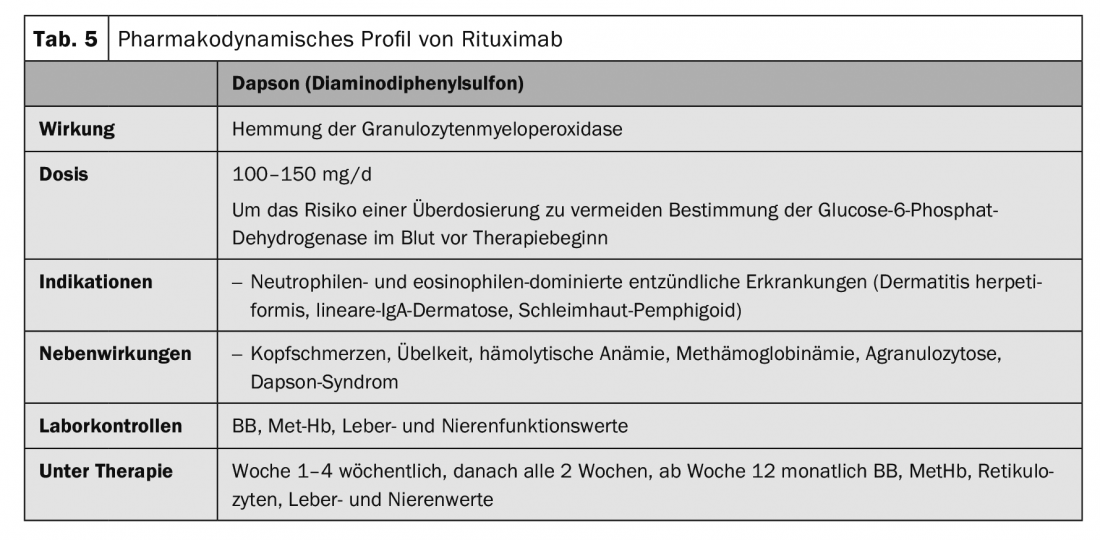
La terapia del penfigoide bulloso debe adaptarse a la actividad de la enfermedad y a las comorbilidades de cada paciente. Por lo general, se utilizan métodos terapéuticos más suaves que para las enfermedades pénfigo. En el penfigoide ampolloso localizado y moderado, los tratamientos desinfectantes y antiinflamatorios tópicos con propionato de clobetasol dos veces al día suelen ser suficientes. Sin embargo, el tratamiento tópico de gran superficie dos veces al día suele ser poco práctico en pacientes de edad avanzada. Suele ser necesario un tratamiento sistémico. La terapia sistémica con un equivalente inicial de 0,5 mg/kg/d de prednisolona en combinación con inmunosupresores adyuvantes como la azatioprina o el micofenolato mofetilo para ahorrar glucocorticoides es el tratamiento de primera elección además del tratamiento tópico (tab. 1). Otras terapias adyuvantes descritas son el metotrexato (15 mg/semana), la dapsona (100 mg/d, tab. 5) y la tetraciclina (200 mg/d). Con la inmunoadsorción (tab. 3) , se pudo demostrar una rápida disminución de los niveles de autoanticuerpos del penfigoide bulloso en el suero y un efecto terapéutico asociado en una serie de casos con 20 pacientes [6].
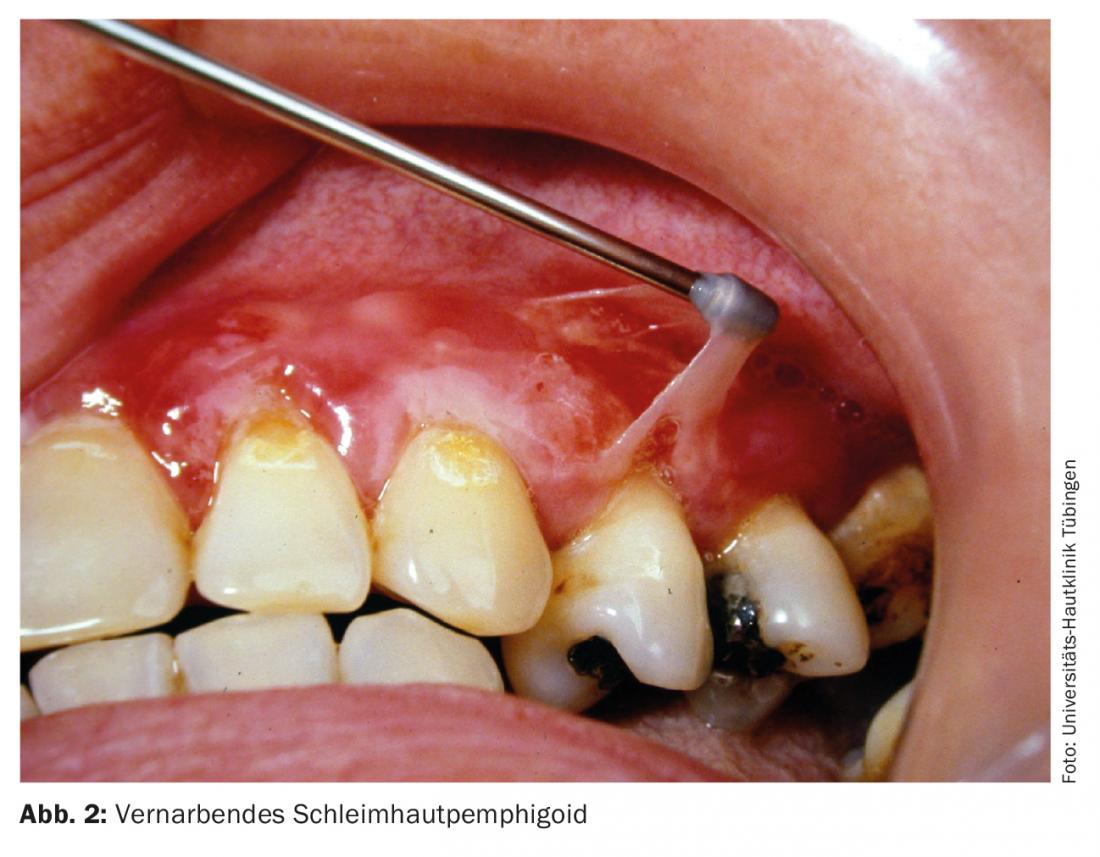
Penfigoide de las mucosas
Esta dermatosis autoinmune también se denomina penfigoide cicatricial. La enfermedad se caracteriza por una gran heterogeneidad clínica con y sin cicatrización. Pueden verse afectadas todas las mucosas con epitelio escamoso (mucosa oral, conjuntiva, nasofaringe, esófago, vulva y recto). La terapia depende de la manifestación clínica; sobre todo, debe evitarse la formación de cicatrices (Fig. 2). Además de altas dosis de glucocorticoides en combinación con inmunosupresores adyuvantes, también puede recomendarse la terapia con ciclofosfamida (tab. 1), IGIV (tab. 4 ) o rituximab (tab. 2) para los pacientes refractarios [7].
Dermatosis IgA lineal
Los agentes de primera línea para las dermatosis IgA lineales son los glucocorticosteroides y la dapsona (tab. 5). Los glucocorticosteroides son mucho menos eficaces en esta enfermedad que en el pénfigo o el penfigoide bulloso. La inmunoadsorción (tab. 3), la IGIV (tab. 4 ) y el rituximab (tab. 2) [8,9] se han descrito como eficaces en informes de casos.
Epidermólisis bullosa adquirida
La epidermólisis ampollosa adquirida muestra una marcada resistencia a la terapia en muchos casos. La forma mecanobullosa en particular suele ser refractaria al tratamiento con glucocorticosteroides en combinación con inmunosupresores adyuvantes como la azatioprina o el micofenolato mofetilo. (Tab.1). En una serie de casos con 3 pacientes y en informes de casos individuales se demostró que Rituximab (Tab. 2) como monoterapia conduce a remisiones duraderas en más de la mitad de los pacientes y en combinación con IGIV ( tab. 4) o inmunoadsorción (tab. 3) en más del 75% de los pacientes [10].
Dermatitis herpetiforme Duhring
Además de la enfermedad cutánea, siempre está presente al mismo tiempo una enteropatía sensible al gluten (enfermedad celíaca), aunque más del 80% de los pacientes no muestran ningún síntoma gastrointestinal, pero suelen presentar atrofia vellositaria subclínica o inflamación en el yeyuno. La terapia se basa en una dieta sin gluten de por vida. Una dieta sin gluten también previene la aparición de linfomas no Hodgkin y probablemente también de carcinomas en el tubo digestivo. El efecto de la dieta sin gluten sobre la piel sólo aparece al cabo de varios meses, por lo que siempre debe utilizarse dapsona (tab. 5) al principio. La enteropatía subyacente no se ve afectada por la dapsona. En caso de intolerancia a la dapsona, puede administrarse sulfasalazina o colchicina.
Resumen
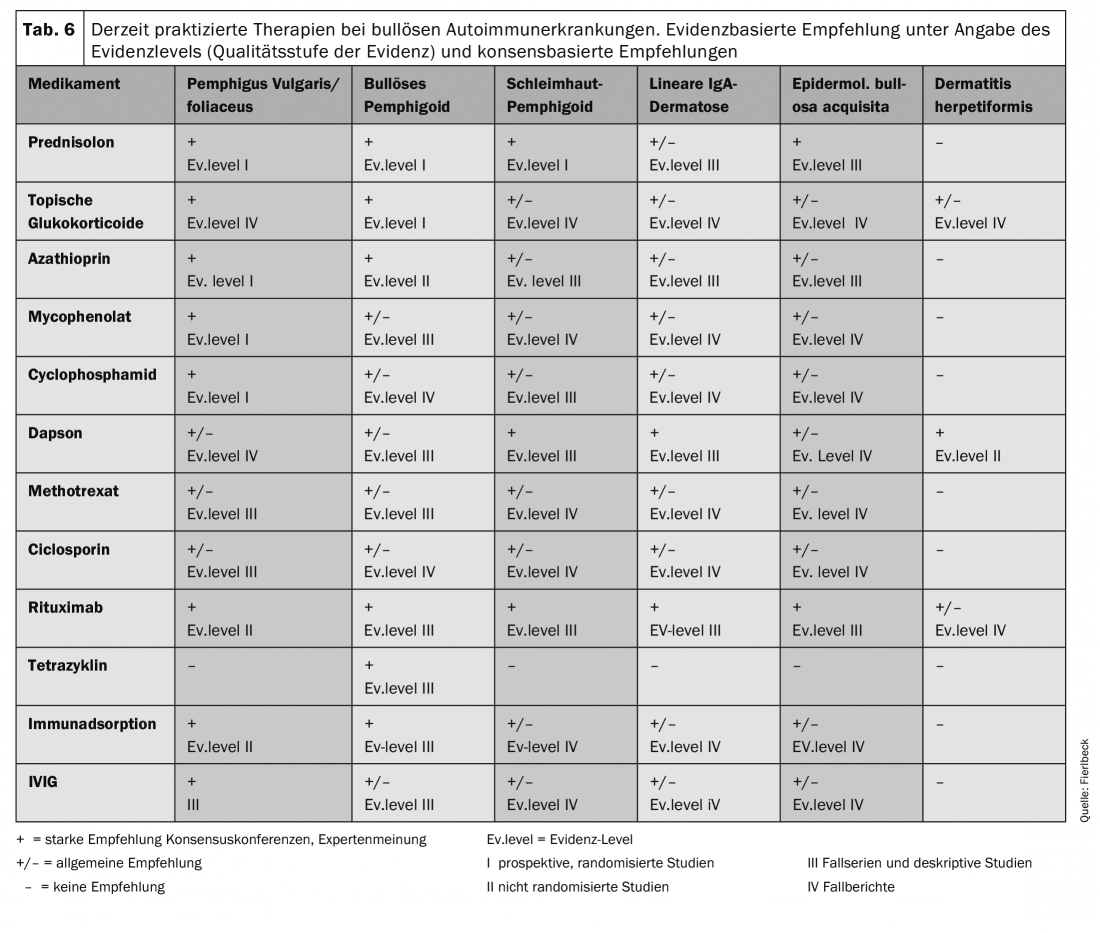
La tabla 6 ofrece una visión general de las terapias practicadas en la actualidad. Las recomendaciones terapéuticas se basan en estudios clínicos (nivel de evidencia) y recomendaciones generales (conferencias de consenso, opiniones de expertos).
Mensajes para llevarse a casa
- Los glucocorticosteroides locales y orales en combinación con la azatioprina son agentes de primera línea, excepto para la dermatitis herpetiforme.
- En el penfigoide ampolloso, puede realizarse un ensayo con glucocorticosteroides tópicos potentes. Sin embargo, suele ser necesario un tratamiento oral con dosis bajas de glucorticosteroides en combinación con azatioprina.
- El rituximab está indicado para las dermatosis ampollosas autoinmunes graves y refractarias. Debido al efecto retardado del rituximab, deben combinarse inicialmente glucocorticosteroides.
- La inmunoadsorción conduce a una mejoría clínica en pocos días; es necesaria una inmunosupresión adicional debido al rápido rebote de los autoanticuerpos y al deterioro clínico.
- En la dermatitis herpetiforme, se requiere una dieta sin gluten de por vida, incluso sin la presencia de una enteropatía manifiesta. La dapsona es el fármaco de primera elección. El efecto de la dieta sin gluten sólo se manifiesta al cabo de meses, la dapsona actúa en pocos días.
Literatura:
- STIKO: Comunicación de la Comisión Permanente de Vacunación (STIKO) en el RKI. Fundamento científico para recomendar la vacunación con la vacuna total de la subunidad del herpes zóster. Instituto Robert Koch (RKI), Boletín epidemiológico 50, 13.12.2018. www.rki.de
- Jolly P, et al: Rituximab de primera línea combinado con prednisolona sola a corto plazo para el tratamiento del pénfigo. Lancet 2017; 389(10083): 2031-2040. dx.doi.org/10.1016/SO140-6736(17)30070-3
- Murrell DF, et al: Diagnóstico y tratamiento del pénfigo: recomendación de un grupo internacional de expertos. J Am Acad Dermatol 2018 Feb 10. pii: S0190-9622(18)30207-X. doi: 10.1016/j.jaad.2018.02.021. [Epub ahead of print]
- Zillikens D, et al: Recomendación para el uso de la inmunoféresis en el tratamiento de la enfermedad ampollosa autoinmune. JDDG 2007; 5(10): 366-373.
- Behzad M, et al: El tratamiento combinado con inmunoadsorción y rituximab conduce a una remisión clínica rápida y prolongada en el pénfigo vulgar. Br J Dermatol 2012; 166(4): 844-852.
- Hübner F, et al: Tratamiento adyuvante del penfigoide ampolloso grave/refractario con inmunoadsorción de proteína A. JDDG 2008; 16(9): 1109-1119.
- Le-Roux-Viller C, et al: Rituximab para pacientes con penfigoide mucoso refractario. Arch Dermatol 2011; 147(7): 843-849.
- Enk A, et al: Uso de altas dosis de inmunoglobulinas en dermatología. JDDG 2009; 7(9): 806-812.
- Pinard C, et al: Dermatosis bullosa IgA lineal tratada con rituximab. JAAD Case Rep. 2019; 5(2): 124-126. doi: 10.1016/j.jdcr.2018.11.004
- Bevans SL, Sami N: El uso de rituximab en el tratamiento de la epidermólisis bullosa adquirida: Tres nuevos casos y una revisión de la literatura. Dermatol Ther 2018; 31(6):e12726. doi: 10.1111/dth12726 [Epub 2018 Oct 3]
PRÁCTICA DERMATOLÓGICA 2019; 29(2): 21-27