Típico de la amiloidosis cardiaca es el desarrollo de insuficiencia cardiaca con fracción de eyección preservada (IC-FEP). Nuevos procedimientos diagnósticos y opciones terapéuticas arrojan ahora una luz diferente sobre esta enfermedad rara, hasta ahora prácticamente incurable.
Con la introducción de nuevos procedimientos diagnósticos y opciones terapéuticas para lo que ha sido una enfermedad rara prácticamente incurable, el interés por la amiloidosis está creciendo rápidamente. La afectación cardiaca (amiloidosis cardiaca) suele provocar una insuficiencia cardiaca grave con todas sus consecuencias clínicas. El objetivo de este artículo es ofrecer una breve visión general del diagnóstico y la terapia modernos de la amiloidosis cardiaca.
Origen
En la amiloidosis sistémica, hay depósitos de proteínas anormalmente plegadas y agregadas en el tejido. Dependiendo de la infestación del órgano, se presentan los correspondientes trastornos funcionales y enfermedades. Si los depósitos se encuentran en el corazón (en el intersticio entre las células miocárdicas), se produce una cardiopatía con el cuadro clínico de la insuficiencia cardiaca. En el corazón suelen depositarse dos tipos diferentes de amiloide, dependiendo de la proteína antecedente defectuosa.
Amiloidosis AL: Suele desarrollarse a partir de una discrasia de células plasmáticas. Las células plasmáticas clonales (por ejemplo, en el mieloma múltiple) producen un exceso de cadenas ligeras libres. En el caso de las cadenas ligeras amiloidógenas, éstas se depositan en forma de fibrillas amiloides en diversos órganos. La afectación cardiaca aislada es rara en la amiloidosis AL. Lo más frecuente es que también se vean afectados los riñones, el hígado y el sistema nervioso periférico. Las cadenas ligeras lambda se detectan con mucha más frecuencia como componentes amiloides que las cadenas kappa.
Amiloidosis ATTR: La transtiretina (TTR) es una proteína transportadora, el 98% de la cual se produce en el hígado y es responsable del transporte de tiroxina y retinol. La TTR es un tetrámero que puede disociarse en cuatro monómeros. Normalmente, estos monómeros son solubles en la sangre. Sin embargo, si existe un trastorno, los monómeros se agregan para formar fibrillas amiloides patológicas. Existen dos causas de amiloidosis TTR: La forma mutada (hereditaria, familiar) se produce debido a una mutación en el gen TTR. La prevalencia de las mutaciones difiere según la región geográfica y los grupos étnicos. La forma “salvaje” (adquirida, senil) no suele manifestarse hasta una edad avanzada y afecta predominantemente al corazón.
Manifestación clínica
Afectación cardiaca: Típico de la amiloidosis cardiaca es el desarrollo de insuficiencia cardiaca con fracción de eyección preservada (ICfpEF). Los depósitos de amiloide engrosan las paredes de los ventrículos y las aurículas (sin hipertrofia real, ya que las células del músculo cardiaco en sí no se ven afectadas). Esto provoca un “endurecimiento” del corazón y una restricción de la relajación (disfunción diastólica). Así, se produce un aumento de las presiones de llenado telediastólico en el ventrículo, una dilatación de las aurículas y un aumento de la presión pulmonar. Clínicamente, esto acaba provocando una insuficiencia cardiaca izquierda y derecha con disnea, intolerancia al rendimiento y edema. También es típico que el ventrículo rígido ya no pueda ajustar correctamente el gasto cardíaco. Debido a que el volumen sistólico está fijado por la rigidez, el gasto cardíaco prácticamente sólo puede ser controlado por la frecuencia cardíaca. Por lo tanto, pueden producirse síncopes y una grave desregulación ortostática, sobre todo en situaciones de estrés, especialmente en pacientes que toman betabloqueantes.
La aurícula izquierda agrandada es un sustrato “bueno” para el desarrollo de la fibrilación auricular. Las presiones elevadas, junto con la escasa movilidad de la aurícula debido a los depósitos de amiloide, pueden provocar un mayor riesgo de trombos intraauriculares y explicar el riesgo significativamente mayor de infartos cerebrales en los pacientes con amiloidosis.
Si los depósitos amiloides afectan al sistema de conducción, se producen bloqueos, especialmente bloqueos AV. Además, aumenta el riesgo de muerte súbita cardiaca debida a arritmias ventriculares (especialmente fibrilación ventricular y “actividad eléctrica sin pulso” – PEA).
Otra manifestación típica de la amiloidosis cardiaca es el dolor torácico sin que se encuentre una enfermedad arterial coronaria real. Por un lado, pequeñas microtrombosis en la microcirculación pueden desencadenar una angina de pecho (espasmos); por otro, la alteración de la función endotelial revela una disfunción microvascular.
Afectación de otros órganos: El frecuente problema de ortostatismo en los pacientes con amiloidosis suele estar causado por daños en el sistema nervioso periférico y la reducción asociada de la vasoconstricción de los vasos periféricos.
Las alteraciones de la inervación intestinal pueden provocar trastornos del tránsito, flatulencias, dolores abdominales e irregularidades en las deposiciones. La afectación renal suele manifestarse por proteinuria y una disminución de la función renal.
Los depósitos de amiloide en el tracto gastrointestinal provocan ocasionalmente malabsorción, pérdida de peso y aumento de la rigidez hepática. En la amiloidosis AL pueden encontrarse ocasionalmente (10%) los signos patognomónicos de hemorragia periorbitaria y macroglosia. La amiloidosis TTR suele ir precedida del síndrome del túnel carpiano.
Diagnóstico
Si se sospecha clínicamente de amiloidosis cardiaca, se utilizan diversos procedimientos diagnósticos. El ECG suele mostrar un bajo voltaje periférico (atenuación de la conducción eléctrica a los electrodos debido al depósito de amiloide), un rasgo distintivo de la miocardiopatía hipertrófica, en la que suele observarse un aumento del voltaje. También pueden verse imágenes de bloqueo, fibrilación auricular y otras arritmias. Sin embargo, todos estos cambios no son específicos de la enfermedad. Además de los parámetros habituales, deben medirse en el laboratorio los biomarcadores cardiovasculares NT-proBNP y troponina. Tampoco debe faltar un examen de orina, especialmente con la cuestión de la albúmina y la orina. La electroforesis de proteínas y la inmunofijación en suero y orina también son esenciales para distinguir las formas AL y ATTR.
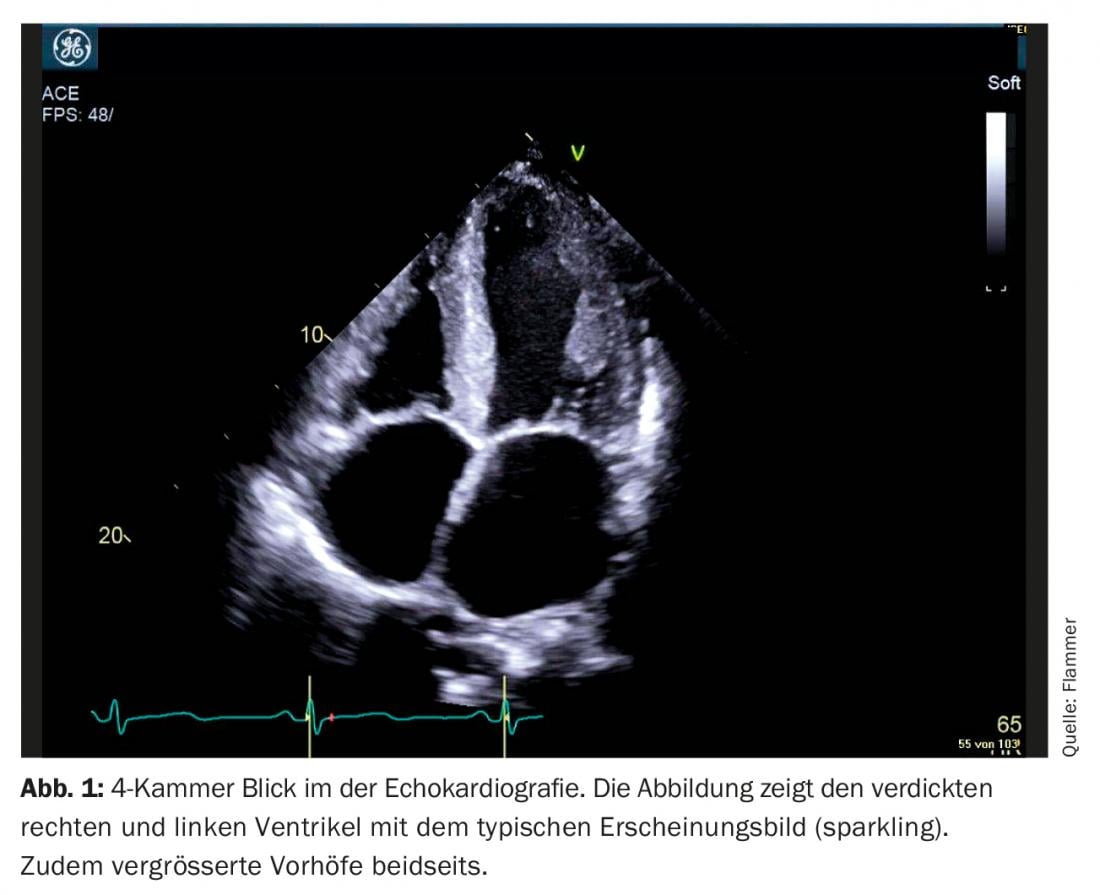
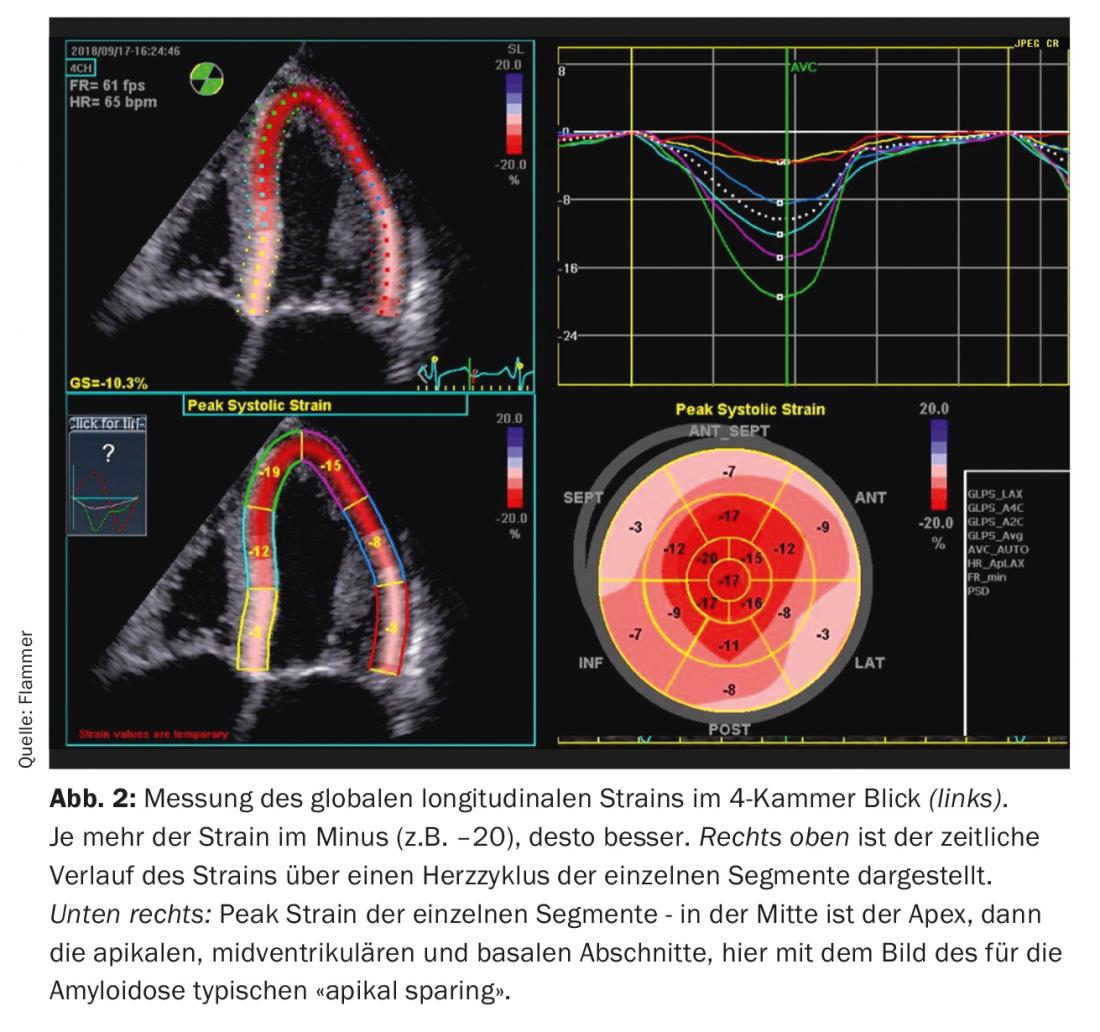
Ecocardiografía: El diagnóstico de la amiloidosis cardiaca es impensable sin la imagen cardiaca. Los pacientes con antecedentes y signos de insuficiencia cardiaca suelen someterse a una ecocardiografía. Si hay signos correspondientes de amiloidosis en la ecografía e insuficiencia cardiaca manifiesta, debe considerarse la amiloidosis. ¿Cuáles son los signos típicos en la ecocardiografía? Hay un miocardio biventricular engrosado, y el tabique intraauricular y las paredes auriculares también suelen estar engrosadas. El miocardio también se presenta específicamente como “centelleante”, lo que es característico de la enfermedad (sin embargo, es importante recordar que la “imagen de segundo armónico” debe estar desactivada en el ecógrafo). El amiloide también se deposita en las válvulas. Así, aparecen ligeramente engrosadas, pero la función de las válvulas suele ser normal. También es frecuente el derrame pericárdico discreto (Fig. 1). Desde el punto de vista funcional, especialmente en la enfermedad avanzada, existe una disfunción diastólica grave, normalmente con un patrón de llenado restrictivo. Debido al aumento asociado de las presiones diastólicas, ambas aurículas se agrandan durante el curso de la enfermedad (dilatación biatrial), característica de la disfunción diastólica grave. Aunque la fracción de eyección suele ser normal, también existe una disfunción sistólica, que se manifiesta en una “deformación” reducida (la deformación describe la deformación medida del miocardio). En particular, la “tensión” longitudinal global es limitada. Típico y específico de la amiloidosis es el llamado “apical sparing”, es decir, la distensión es normal (spared) en el ápex pero se deteriora cuanto más se avanza hacia la base del corazón (Fig. 2).
Resonancia magnética (RM): Hoy en día, la amiloidosis se diagnostica a menudo mediante este examen (a veces como hallazgo incidental en otras investigaciones). Los cambios observados en la resonancia magnética son similares en principio a los observados en la eco, pero pueden apreciarse más cambios típicos, sobre todo si se utiliza también el medio de contraste gadolinio. Debido al lento lavado del gadolinio, éste permanece más tiempo en el intersticio si hay amiloide. La resonancia magnética muestra entonces un “realce tardío del gadolinio” difuso y parcheado, típico de la amiloidosis.
Algoritmo diagnóstico para la sospecha de amiloidosis cardiaca
En pacientes con un cuadro clínico correspondiente y sospecha por imagen de amiloidosis cardiaca, debe descartarse ante todo una discrasia de células plasmáticas. Para ello, hay que recurrir a la electroforesis de proteínas y a la inmunofijación en la orina y el suero, como se ha descrito anteriormente. Si hay alguno presente, se busca principalmente la amiloidosis AL y, en ausencia de signos, la amiloidosis ATTR (Fig. 3).
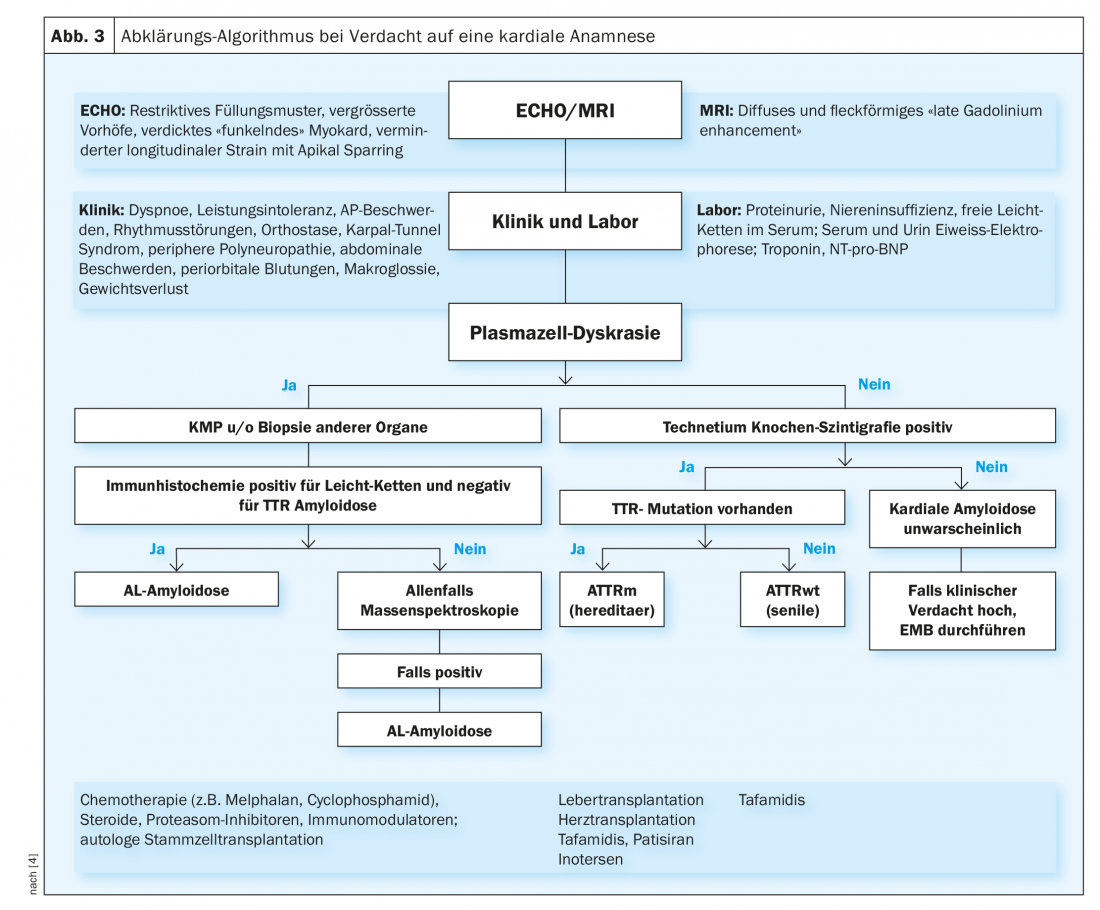
Si se sospecha de amiloidosis AL (discrasia de células plasmáticas presente), debe intentarse detectar directamente el amiloide AL biotópicamente. Normalmente se realiza un aspirado de médula ósea para diagnosticar la enfermedad hematológica subyacente (normalmente mieloma múltiple). Dado que la amiloidosis es una enfermedad sistémica, el diagnóstico se confirma en cuanto se detecta amiloide AL en una biopsia (de cualquier tejido). La menos invasiva y traumática es la aspiración del tejido adiposo abdominal. Asimismo, a veces puede buscarse amiloide en muestras de tejido tomadas previamente como parte de una colonoscopia o una gastroscopia. Además, pueden realizarse biopsias de glándulas salivales, labio o recto. En ocasiones, también es necesaria una biopsia renal o endomiocárdica para realizar un diagnóstico definitivo. En la amiloidosis cardiaca, la biopsia endomiocárdica es muy sensible. Por un lado, se examinan las biopsias en busca de amiloide y, por otro, se utiliza la inmunohistoquímica para intentar clasificar el tipo de amiloidosis. En los casos difíciles, es necesario recurrir a la espectroscopia de masas (patrón oro). Sin embargo, sólo en unas pocas patologías se lleva a cabo un examen de este tipo.
Si se sospecha una amiloidosis TTR (falta de evidencia de discrasia de células plasmáticas), hoy en día se realiza una gammagrafía con tecnecio (ósea). En estas condiciones, la sensibilidad y especificidad de esta metodología en la amiloidosis ATTR cardiaca es muy buena. Si los resultados son positivos, puede omitirse la biopsia y establecerse el diagnóstico. Si la gammagrafía no es clara y se sigue sospechando una amiloidosis cardiaca, debe realizarse una biopsia endomiocárdica en este lugar.
Una vez confirmada la amiloidosis ATTR, es necesario realizar pruebas genéticas para distinguir si se trata de una forma “salvaje” o hereditaria.
Terapia de la amiloidosis cardiaca
La insuficiencia cardiaca suele estar en el centro de los síntomas de la amiloidosis cardiaca. Desde el punto de vista terapéutico, deben abordarse ante todo. Los diuréticos ayudan sintomáticamente. Sobre todo porque las presiones de llenado cardiaco están elevadas debido a la fisiología cardiaca restrictiva, lo que provoca una elevación de la presión pulmonar venosa pasiva. Los pacientes son significativamente menos sintomáticos bajo diuréticos. En el curso de la enfermedad suelen necesitarse dosis cada vez más altas, por lo que también es importante asegurarse de que se sustituye por una cantidad suficiente de potasio. La titulación de la dosis suele ser un reto (recuadro).

Según la fisiología de la IC-FEM, no hay indicación para los inhibidores de la ECA, los betabloqueantes y los antagonistas de la aldosterona. Por el contrario, los inhibidores de la ECA en particular pueden exacerbar lo que a menudo ya es una ortostatismo pronunciado. Los betabloqueantes provocan la incapacidad de aumentar el gasto cardíaco en situaciones de estrés debido al volumen sistólico fijo. Así pues, los betabloqueantes se suspenden en pacientes con fibrilación auricular taquicárdica.
La fibrilación auricular y los infartos cerebrales resultantes son frecuentes. Por un lado, es esencial no pasar por alto la FVH (exámenes Holter semestrales), por otro, debe iniciarse la anticoagulación oral si está presente, independientemente de la puntuación CHADS-Vasc. No está del todo claro si debe iniciarse el OAK en ausencia de FVC – es probable que sea apropiado en el caso de un patrón de llenado restrictivo. En la FHV taquicárdica, debe realizarse un control de la frecuencia con betabloqueantes, no debe administrarse digoxina debido a su mayor toxicidad. La amiodarona está indicada en ciertos pacientes para el control del ritmo y ocasionalmente para el control de la frecuencia.
Debido a las alteraciones en el sistema de conducción cardiaca, se hace necesaria la implantación de marcapasos, especialmente en pacientes con amiloidosis TTR, una indicación que puede darse con generosidad. La cuestión de si implantar o no un desfibrilador cardioversor implantable (DCI) es especialmente difícil. La causa más común de muerte cardiaca en pacientes con amiloidosis es la muerte cardiaca súbita debida a arritmias ventriculares. No sólo se observa fibrilación ventricular, sino también un número relevante de PEA. Estos últimos no son detectados de forma fiable por un DCI. También hay una mayor tasa de descargas aplicadas incorrectamente (lo que resulta muy traumático para los pacientes) y de descargas fallidas. Hasta ahora, sólo existen estudios retrospectivos sobre este tema, que no muestran ningún beneficio claro para un DAI. Por lo tanto, la indicación debe hacerse con cautela y discutirse en detalle en un equipo de amiloidosis.
Terapia causal
En la amiloidosis AL, la enfermedad hematológica debe abordarse en primer lugar. Sin embargo, esto es a menudo un reto, especialmente en casos de afectación cardiaca grave e insuficiencia cardiaca grave concomitante. La terapia de altas dosis con trasplante autólogo de células madre, a la que a menudo se aspira, no puede llevarse a cabo en este caso. Los otros regímenes terapéuticos con agentes quimioterapéuticos (por ejemplo, melfalán, ciclofosfamida), esteroides y nuevos inhibidores del proteasoma, inmunomoduladores o anticuerpos anti-CD38 también pueden ser ocasionalmente estresantes para el corazón. Sin embargo, existen enfoques curativos en algunos casos. Sin embargo, un tratamiento detallado iría más allá del alcance de este artículo.
En el caso de la amiloidosis TTR hereditaria, el trasplante de hígado (y de corazón en fases avanzadas) sigue siendo el tratamiento de elección.
Sin embargo, desde el verano de 2018, hay dos fármacos disponibles en EE.UU. y Europa (aprobados por la FDA y la EMA) que utilizan la tecnología del ARN de interferencia para “silenciar” el gen defectuoso de modo que no se produzca TTR amiloidogénica. Ninguno de los dos fármacos ha sido aún aprobado en Suiza. Un estudio a gran escala en pacientes con amiloidosis hereditaria demostró de forma impresionante cómo puede detenerse la polineuropatía (que suele ser frecuente en la amiloidosis hereditaria). Los análisis de subgrupos del fármaco patisiran muestran que los resultados en el corazón podrían ser igualmente buenos, pero aún quedan por ver más estudios. Hasta hace poco, no existía una terapia causal para la amiloidosis TTR de tipo salvaje. Afortunadamente, un estudio publicado también en el verano de 2018 demostró que el tafamidis, un estabilizador de la transtiretina, que también está aprobado en Europa y EE.UU. para el tratamiento de la neurpatía amiloide hereditaria, tiene buenos resultados en la amiloidosis cardiaca con mejora de la calidad de vida, reducción de la mortalidad y reducción de las hospitalizaciones relacionadas con el corazón. El fármaco aún no está disponible en Suiza, pero se espera su aprobación en breve debido a los buenos resultados.
Mensajes para llevarse a casa
- La amiloidosis AL es una enfermedad rara que suele desencadenarse por una enfermedad hematológica subyacente “productora de cadenas ligeras” y suele afectar a otros órganos además del corazón. En el centro de la terapia está el tratamiento de la enfermedad hematológica.
- La amiloidosis TTR puede estar causada por una mutación en el gen TTR o puede ser “adquirida” en el transcurso de la vida (“de tipo salvaje”). La forma familiar, más rara, se manifiesta predominantemente con polineuropatía o cardiopatía (o una combinación), dependiendo del gen implicado; la forma “salvaje” suele limitarse al corazón. Además de la terapia sintomática, existen nuevos enfoques terapéuticos prometedores con estabilizadores de la TTR y moléculas de ARN de interferencia.
- El diagnóstico de la amiloidosis cardiaca requiere una clínica adecuada (insuficiencia cardiaca), hallazgos en la ecografía o la resonancia magnética típicos de la amiloidosis y pruebas de amiloide en una biopsia o una gammagrafía con Tc positiva.
- El cuadro clínico típico de la amiloidosis cardiaca incluye, en particular, síntomas de insuficiencia cardiaca, ortostatismo, síncope, arritmias (especialmente HFV y taquicardia ventricular/fibrilación ventricular), bloqueo AV y angina microvascular.
Literatura:
- Brouwers S, et al: Cardiac amyloidosis Cardiovasc Med. 2018; 21(11): 282-289.
- Rauch PJ, et al: Amiloidosis sistémicas Switzerland Med Forum 2014 14; 943-948.
- Laptseva N, et al: Amiloidosis cardiaca: todavía un reto Eur Heart J 2017, 38(22): 122.
- Falk RH, et al: Amiloidosis cardiaca AL (cadena ligera): revisión del diagnóstico y la terapia.
- Gillmore JD, et al: Diagnóstico sin biopsia de la amiloidosis cardiaca por transtiretina Circulation 2016: j133(24): 2404-2412.
- Maurer MS, et al: Tratamiento con tafamidis para pacientes con miocardiopatía amiloide por transtiretina N Engl J Med 2018; 379(11): 1007-1016.
- Adams D, et al: Patisiran, una terapia de ARNi, para la amiloidosis hereditaria por transtiretina N Engl J Med 2018; 379(1): 11-21.
CARDIOVASC 2019; 18(2): 6-10