Los síntomas psiquiátricos son un aspecto crucial de la enfermedad de Huntington. Una visión general de la evaluación de los síntomas y las opciones de tratamiento.
Los síntomas psiquiátricos son un aspecto crucial de la enfermedad de Huntington. También suelen aparecer en la fase prodrómica y empeoran con la progresión de la enfermedad [1].
La enfermedad de Huntington, que tiene una prevalencia de 10,6-13,7/100.000 habitantes en el mundo occidental, se basa en un trastorno poliglutamínico autosómico dominante causado por una elongación de la repetición CAG en el gen Huntington del cromosoma 4 [2,3]. Cuantas más repeticiones, mayor será la gravedad y menor el periodo de latencia hasta la primera manifestación de los síntomas. La enfermedad se manifiesta clínicamente a partir de una longitud de 39 repeticiones [4]. Esto provoca la degeneración de las neuronas gaba y colinérgicas en el neoestriado y la atrofia del cuerpo estriado.
Los síntomas neuropsiquiátricos pueden dividirse en cuatro subáreas: Impulso, afecto, delirio y cambios de personalidad [5]. Pueden aparecer comportamientos compulsivos, depresión, síntomas psicóticos, confabulaciones y desarrollo de demencia. Son frecuentes las alteraciones de la flexibilidad cognitiva y del control de los impulsos [6].
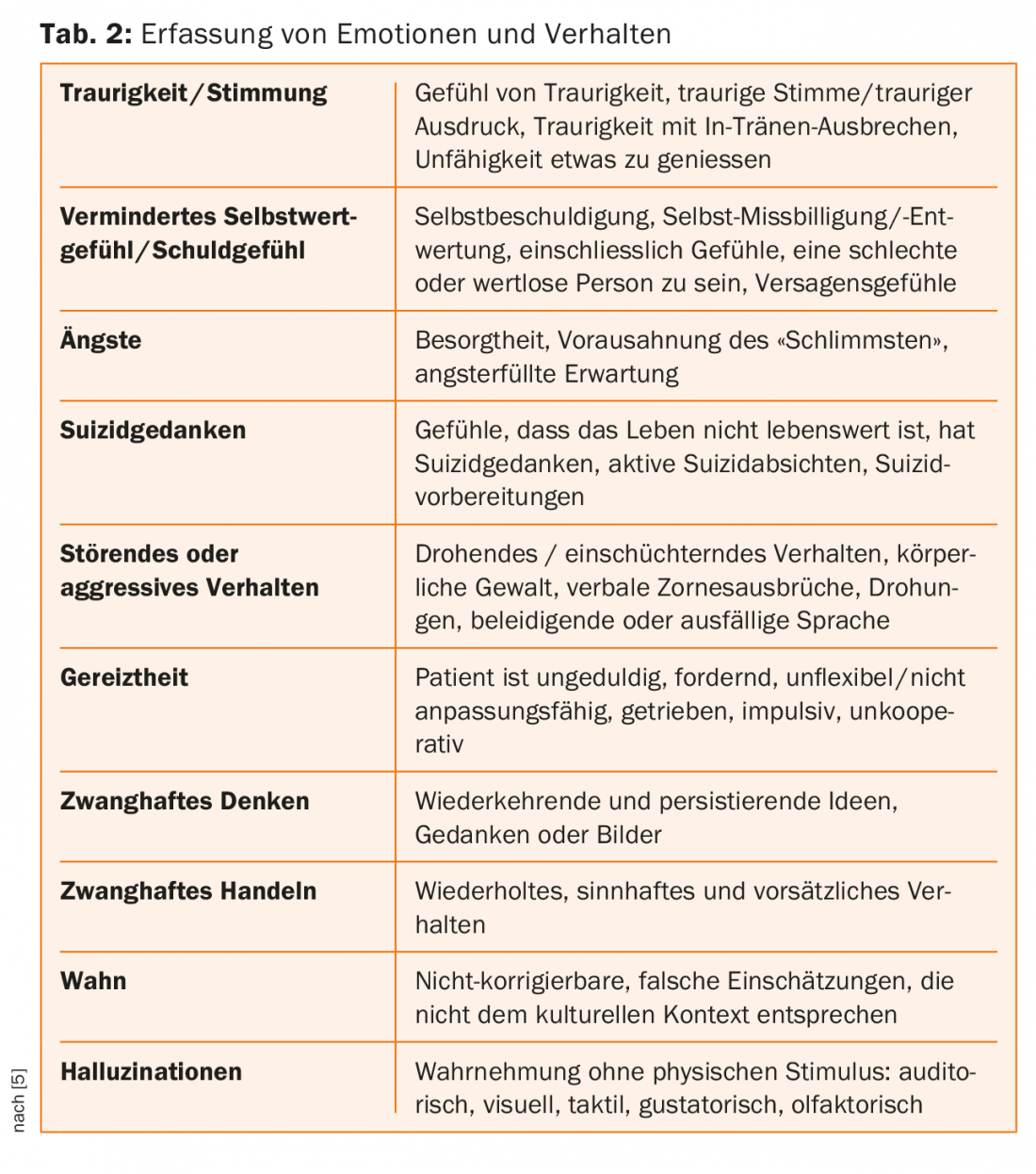
Junto con el registro del comportamiento, la calidad de vida y la capacidad de funcionamiento del paciente, los síntomas psiquiátricos se evalúan en la “Escala unificada de calificación de la enfermedad de Huntington” (UHDRS), estándar de examen, con calificaciones en una escala de Likert de 0 a 4 respecto a su gravedad [5] – ver Tabla 1. Además de la depresión, también puede observarse un aumento de la irritabilidad en el curso de la enfermedad; a menudo se encuentra ya en la fase prodrómica o de manifestación precoz. La perspicacia habitualmente ausente en la enfermedad se atribuye al deterioro de la función en el lóbulo frontal y a la reducción de la transmisión hacia el cuerpo estriado [7].

Sin embargo, estos síntomas psiquiátricos también pueden aparecer en otras enfermedades neurológicas o psiquiátricas, por lo que el diagnóstico precoz de la enfermedad de Huntington sobre la base de los síntomas psiquiátricos es difícil [1].
Los síntomas motores típicos son los movimientos coreaticos: contracciones musculares involuntarias, arrítmicas y cortas que se producen en regiones cambiantes del cuerpo. Además, hay síntomas vegetativos como alteraciones del sueño, pérdida de peso y cambios en la libido [6]. En la variante clásica de la enfermedad, aparecen precozmente -además de los movimientos coreaticos- trastornos de los movimientos oculares, distonía e impersistencia motora al realizar acciones motoras simples. Las deficiencias motoras reducen la esperanza de vida de los pacientes, entre otras cosas debido a las caídas y a la neumonía por aspiración [2].
Historial médico y estado
El psicólogo que la trataba asignó a la paciente, de 42 años, una terapia de hospitalización. En el entorno familiar, habían aumentado los conflictos debido a un cambio en la naturaleza del paciente que se percibía desde el exterior. Esto ya había comenzado hacía cinco años, y la historia externa de la paciente la mostraba cada vez más irritable, verbalmente agresiva y cada vez menos capaz de sobrellevar la situación. Es propensa a los arrebatos emocionales impulsivos y hace insinuaciones incomprensibles a los miembros de su familia. La paciente se había vuelto “más desconfiada” y se había retraído socialmente. Por la mañana, tiene cada vez más dificultades para levantarse y está apática. Había dejado de cumplir con sus obligaciones domésticas y educativas y en ocasiones había mostrado un “comportamiento extraño”. El marido se había mudado debido al deterioro de la situación, los dos hijos comunes vivían con la cuñada. La propia paciente expresó que tenía la sensación de que su cuñada quería quitarle a sus hijos.
Como consecuencia de un grave accidente de bicicleta sufrido hace 18 años, recibe una pensión de invalidez. Por lo demás, no había enfermedades previas físicas o psiquiátricas conocidas, el historial de adicciones era anodino.
La paciente no conocía su enfermedad y apenas tenía motivación para el tratamiento. En el momento del ingreso, presentaba un discreto trastorno involuntario de la movilidad, que la paciente declaró no haber notado por sí misma. Cuando se le preguntó, el marido informó de un aumento de los trastornos del movimiento de los brazos y las manos, así como del tronco. No se conocen enfermedades psiquiátricas o neurológicas en la familia.
Hallazgos clínicos
Los hallazgos psicopatológicos mostraron una reducción de la atención, la memoria y la concentración. El tren de pensamiento formal se ordenó, el pensamiento se estrechó. La paciente parecía desconfiada, irritable y melancólica, inestable en el afecto, fácilmente desviable hacia el polo depresivo. Hubo un delirio paranoide con respecto a la cuñada, no hay pruebas de delirios sensoriales ni de trastornos del yo. Aparte de una inquietud motora, la paciente era psicovegetativamente discreta. No había indicios de peligro agudo para sí mismo o para los demás. Neurológicamente, además de los discretos movimientos coreaticos, era impresionante el estado residual tras un accidente de tráfico y una reconstrucción plástica (plástica del dorsal ancho) de la parte inferior de la pierna con paresia y alteraciones sensitivas debidas a una parálisis peronea postraumática. Había ligeras alteraciones en la motricidad fina y la coordinación, así como un patrón de marcha inestable y asimétrico con una cojera rígida. Se informó de un aumento de las sensaciones de presión, temperatura y dolor en la parte inferior de la pierna izquierda.
Diagnóstico y evolución clínica
Un buen mes después de su ingreso en domo, se hizo una presentación en la clínica neurológica del Hospital Cantonal de Lucerna ante la sospecha de un diagnóstico de enfermedad de Huntington. Allí, el examen físico reveló apraxia de la mirada, habla staccato intermitente y discinesias de la boca, la mandíbula y los ojos, así como mioclonía del ángulo oral y discinesias miocloniformes de los dedos bilateralmente. Además, había impersistencia motora de la lengua y las manos (“agarre de lechera”), disdiadococinesia y movimientos coreicos intermitentes, así como un aumento de los reflejos musculares de todas las extremidades y un reflejo cloniforme del tendón de Aquiles bilateral. La ataxia de la postura y de la marcha con un aumento de la discinesia era evidente.
Los exámenes realizados EEG, hiperventilación y fotoestimulación así como la ecografía dúplex de los vasos cerebrales no mostraron ningún resultado patológico. Una RMNc realizada demostró atrofia caudada bilateral sin alteraciones de señal y atrofia putaminal leve.
Esto reforzó la sospecha de la enfermedad de Huntington; se consideraron como diagnósticos diferenciales la discinesia tardía debida a anestésicos antiguos y la enfermedad de Wilson.
En la clínica psiquiátrica, se utilizaron 300 mg de quetiapina para tratar los síntomas afectivos y delirantes, y también se integró a la paciente en terapias orientadas a la acción.
Dos meses después de su ingreso, la paciente se retiró a petición propia y tras consultar con el servicio de neurología, mientras seguía medicándose con Quetiapina retard 300 mg. El día del alta se realizó un examen genético molecular en el Hospital Cantonal de Lucerna, que aportó pruebas de una expansión CAG patológica con 46 repeticiones CAG y, por tanto, el diagnóstico confirmado de enfermedad de Huntington.
A pesar de los síntomas mentales relevantes para el tratamiento, el tratamiento antidiscinético aún no era necesario debido a los síntomas motores leves al alta. Además del asesoramiento genético y psicológico, se recomendó la atención posthospitalaria por parte de un servicio social y la atención de proximidad.
Debate
La enfermedad de Huntington es un trastorno neuropsiquiátrico que se diagnostica fácilmente en las últimas fases debido a los movimientos coreaticos (ver Tab. 1-3 para registrar los problemas de comportamiento). Sin embargo, las anomalías neuropsiquiátricas inespecíficas suelen aparecer años antes de la manifestación inicial de los síntomas motores y afectan gravemente a la vida de los pacientes. Por lo tanto, es aún más importante incluir esta “enfermedad huérfana” en el diagnóstico diferencial de los pacientes con anomalías psiquiátricas para poder acompañarlos y atenderlos adecuadamente [8]. En nuestro caso, la irritabilidad paranoide-desconfiada, la falta de control de los impulsos, así como el estado de ánimo depresivo, además de los movimientos coreaticos que se encontraban en los estadios iniciales, estaban a la cabeza.
Hasta ahora no hay posibilidades de una terapia causal, pero se está investigando mucho sobre la mejor manera de influir en la progresión de la enfermedad [9]. Las primeras investigaciones de laboratorio sugieren posibilidades terapéuticas en un futuro próximo [10]. Se habla de transbordadores genéticos, biomarcadores o trasplantes de células madre como posibles terapias curativas, pero aún no son aplicables, por lo que el diagnóstico sigue considerándose incurable [9].
Tras recibir el diagnóstico, el paciente necesita una atención y un asesoramiento integrales. Además de las terapias funcionales (ocupacional y fisioterapia, logopedia), el inicio de la terapia farmacológica está indicado en presencia de síntomas mentales que requieran tratamiento – como en el caso descrito, por ejemplo, con fármacos estabilizadores afectivos y antipsicóticos como los neurolépticos atípicos. En caso necesario, puede administrarse tratamiento con tetrabenazina o tiaprida para la hipercinesia disruptiva y L-dopa para los síntomas parkinsonoides. El asesoramiento genético, así como el apoyo social y psicoterapéutico a los pacientes afectados y a sus familias, son de gran importancia.

Mensajes para llevarse a casa
- La enfermedad de Huntington puede causar síntomas neuropsiquiátricos debilitantes durante la fase prodrómica o en las primeras etapas.
- Los síntomas neuropsiquiátricos proceden de los cuatro subdominios del impulso, el afecto, el delirio y los cambios de personalidad.
- A menudo, el paciente no es consciente de la enfermedad, aunque sus familiares y su entorno sufran.
- Puede realizarse una evaluación de los síntomas, por ejemplo, registrando las anomalías de comportamiento en la “Escala unificada de valoración de la enfermedad de Huntington” (UHDRS).
- Los antipsicóticos atípicos y el apoyo psicoterapéutico pueden utilizarse en el tratamiento de los síntomas neuropsiquiátricos.
Literatura:
- Epping EA, Kim JI, et al: Síntomas psiquiátricos longitudinales en la enfermedad de Huntington prodrómica: una década de datos. Am J Psychiatry. 2016; 173(2): 184-192.
- Pringsheim T, Wiltshire K, et al: La incidencia y prevalencia de la enfermedad de Huntington: una revisión sistemática y metaanálisis. Mov Disord. 2012; 27(9): 1083-1091.
- McColgan P, Tabrizi SJ: Enfermedad de Huntington: una revisión clínica. Eur J Neurol. 2018; 25(1): 24-34.
- Paulson HL, Albin RL: Enfermedad de Huntington: Características clínicas y vías terapéuticas. En: Neurobiología de la enfermedad de Huntington: Aplicaciones al descubrimiento de fármacos (eds Lo DC, Hughes RE): 2011.
- Kieburtz K: Escala unificada de valoración de la enfermedad de Huntington: fiabilidad y consistencia. Trastornos del movimiento. 1996 II(2): 136-142.
- Duff K, Paulsen JS, et al: Predecir el HDIotHSG. Síntomas psiquiátricos en la enfermedad de Huntington antes del diagnóstico: el estudio predict-HD. Biol Psiquiatría. 2007; 62(12): 1341-1346.
- Hoth KF, Paulsen JS, et al: Los pacientes con enfermedad de Huntington tienen alterada la conciencia de sus capacidades cognitivas, emocionales y funcionales. J Clin Exp Neuropsychol. 2007; 29(4): 365-376.
- Schiefer J, Werner C, et al: Diagnóstico clínico y manejo en la enfermedad de Huntington precoz: una revisión. Enfermedades neurológicas y neuromusculares degenerativas. 2015; (5): 37-50.
- Lo DC, Hughes RE, editores. Neurobiología de la enfermedad de Huntington: Aplicaciones al descubrimiento de fármacos. CRC Press/Taylor & Francis, 2011.
- Chen GL, Ma Q, et al.: Modulación del REST nuclear por el splicing alternativo: una posible diana terapéutica para la enfermedad de Huntington. J Cell Mol Med. 2017; 21(11): 2974-2984.
Agradecimientos: Queremos expresar nuestro más sincero agradecimiento al Prof. Dr. med. Bohlhalter y al Dr. med. Stephan Mittas de Neurología Lucerna, al Dipl. med. Berennen-Dietrich del Rötgeninstitut Luzern y el Dr. med. Roland Spiegel de Genetica Zurich.
InFo NEUROLOGÍA Y PSIQUIATRÍA 2018; 16(1): 24-27.