Para el pronóstico de esta enfermedad hereditaria autosómica recesiva, el diagnóstico en el periodo neonatal y el posterior inicio del tratamiento son cruciales, sobre todo en lo que respecta a la aparición de complicaciones como los carcinomas hepatocelulares y el deterioro neurocognitivo. Actualmente se dispone de nitisinona como terapia farmacológica, y también se recomiendan ciertas medidas dietéticas.
En la tirosinemia hepatorrenal tipo 1 (HT1), la formación de la enzima fumarilacetoacetasa (FAH), la última enzima en la degradación de la tirosina, está alterada desde el nacimiento. Esto se debe a una variante patogénica bialélica en el gen FAH. Debido a la deficiencia enzimática, los metabolitos tóxicos fumarilacetoacetato, maleilacetoacetato y succinilacetona se acumulan en el organismo y dañan el hígado, los riñones y el sistema nervioso periférico. Como consecuencia, a menudo se produce una insuficiencia hepática no tratada en el primer año de vida o, en un curso más lento y crónico, se desarrolla una cirrosis hepática o un carcinoma hepatocelular (CHC) [1]. La nitisinona es una sustancia activa que interviene en la cascada de degradación temprana de la tirosina inhibiendo de forma competitiva la enzima 4-hidroxifenilpiruvato dioxigenasa. Esto evita la formación de productos intermedios tóxicos. El tratamiento de todos los genotipos de la enfermedad debe iniciarse lo antes posible para prolongar la supervivencia global y prevenir las manifestaciones orgánicas potencialmente mortales. En Suiza, la nitisinona (Nityr®) se autorizó en forma de comprimidos en 2022 [2]. Iniciar el tratamiento en las primeras semanas de vida no sólo previene el CHC en la mayoría de los casos, sino también la disfunción hepática y renal [3–5].
Antes de la era de la nitisinona, la historia natural de la HT1 solía ser fatal, el 90% de los pacientes con HT1 morían en los dos primeros años de vida y la única opción era el trasplante de hígado [1]. Así pues, la disponibilidad de la nitisinona (NTBC) ha cambiado fundamentalmente el curso clínico y el resultado de las personas afectadas por HT1, según los autores de la directriz S2k sobre el diagnóstico y el tratamiento de la tirosinemia hepatorrenal (tirosinemia de tipo 1), que se actualizó en 2022 [1].
Se recomienda el cribado neonatal
El factor decisivo para el pronóstico a largo plazo, en particular la prevención del CHC, es iniciar el tratamiento en el periodo neonatal. Dado que los primeros síntomas no aparecen en la mayoría de los pacientes hasta que tienen varios meses de edad, el diagnóstico precoz no es posible por motivos puramente clínicos [3]. Por lo tanto, en caso de sospecha clínica, la succinilacetona (SA) debe determinarse cuantitativamente a partir de sangre seca, suero/plasma y/u orina (“cribado selectivo”). La determinación de la concentración de tirosina por sí sola tiene una sensibilidad y especificidad insuficientes, por lo que no se recomienda [1]. Un valor de medición de SA por encima de un punto de corte definido se considera un resultado de cribado positivo. Puede realizarse un análisis genético molecular del gen FAH para confirmar el diagnóstico (recuadro). Si el gen FAHes normal, la directriz recomienda analizar el gen GSTZ1 [1].
| Análisis genéticos moleculares La identificación de variantes patogénicas bialélicas en el gen de la fumarilacetoacetasa (FAH) confirma el diagnóstico de tirosinemia de tipo 1 [1]. El ADN de las células sanguíneas o de otros tejidos corporales (hisopo de mejilla) puede utilizarse para este análisis. También es posible el análisis de ARN a partir de células sanguíneas, muestras de biopsia hepática o fibroblastos cultivados. El análisis de un solo gen de los 14 exones codificantes del gen FAHen pacientes con un cuadro clínico/bioquímico claro es un método establecido desde hace tiempo que detecta variantes patogénicas con una sensibilidad estimada de >95% [1]. Hoy en día, sin embargo, las técnicas de NGS(secuenciación de próxima generación) se utilizan sobre todo como parte de los diagnósticos de panel o como parte de la WES (secuenciación del exoma completo) para el análisis del gen de la FAH, ambas para diagnósticos más rápidos y rentables [16]. |
Iniciar la terapia con nitisinona en las primeras semanas de vida
Los efectos de la nitisinona se basan en la inhibición de la enzima 4-hidroxifenilpiruvato dioxigenasa, implicada en la degradación normal de la tirosina (Fig. 1). Esto impide la formación de metabolitos tóxicos. Los efectos secundarios son raros; los más frecuentes son trastornos del hemograma, trastornos oculares y un aumento de la concentración de tirosina. La interrupción o interrupción brusca del tratamiento con nitisinona puede provocar “crisis de porfiria” y debe evitarse [1, 6-8]. El valor diana terapéutico de la nitisinona aún no está bien definido; en la literatura especializada, se da 20-60 μM como valor diana terapéutico para las concentraciones de nitisinona en plasma [3,9–11]. Según los autores de las directrices, también son posibles concentraciones significativamente más bajas sin perjudicar el control metabólico medido por la supresión de la producción de SA [1]. El intervalo terapéutico objetivo para la concentración de tirosina en sangre se da como 200-800 μM, sin que existan estudios controlados, aleatorizados y comparativos al respecto [3,9–12].
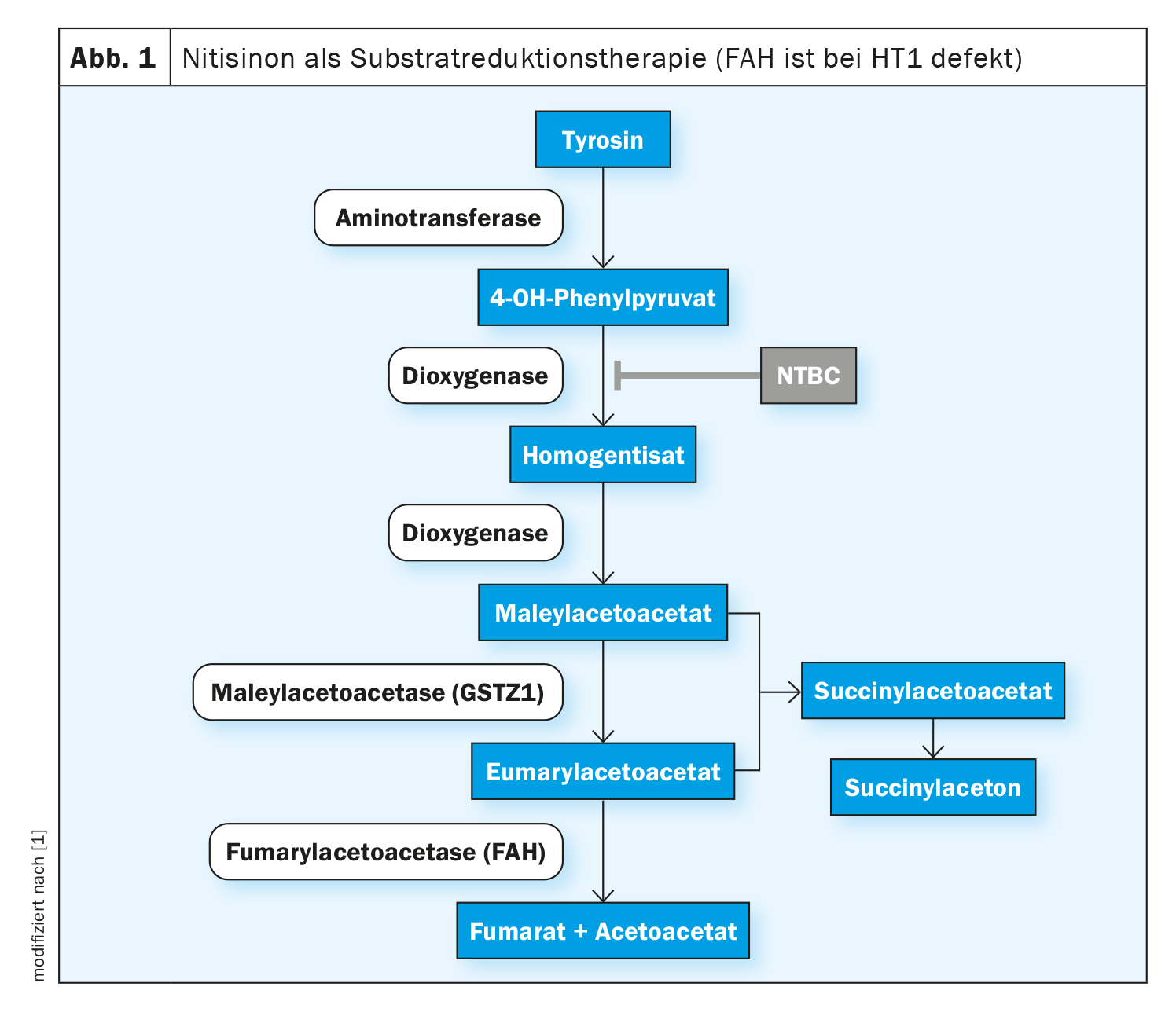
Si es posible, la terapia con nitisinona debe combinarse con una dieta reducida en proteínas suplementada con una mezcla de aminoácidos sin tirosina ni fenilalanina [13–15].
Errores de diagnóstico: los análisis genéticos pueden ayudar
El diagnóstico diferencial más importante de una concentración elevada de SA es la deficiencia de maleil acetoacetato isomerasa, una anomalía metabólica presumiblemente benigna que no se asocia a disfunción hepática [1]. La directriz también señala que las concentraciones de SA ligeramente elevadas -dependiendo del valor de corte utilizado- también pueden ser temporales o deberse a formas leves de la enfermedad que no requieren tratamiento [1]. Un resultado de cribado positivo debe confirmarse mediante uno o más métodos de análisis alternativos además del control en el mismo material de muestra (sangre seca). Esto incluye el análisis cuantitativo por cromatografía de gases/espectrometría de masas (GC/MS) de ácidos orgánicos en orina y la determinación de SA en sangre. Si existe una fuerte sospecha, también es aconsejable comprobar los parámetros de la función hepática. Los hallazgos anormales en los diagnósticos de confirmación pueden aclararse aún más mediante pruebas genéticas moleculares del gen FAH. Si no se detecta, puede analizarse el gen GSTZ1 (glutatión S-transferasa zeta 1-1), cuya deficiencia es la causa de la deficiencia de maleil acetoacetato isomerasa [1].
Literatura:
- “Directriz S2k: Diagnóstico y tratamiento de la tirosinemia hepatorrenal (tirosinemia tipo 1)”, número de registro AWMF: 027-003, a 09.06.2022.
- Swissmedic: Información sobre medicamentos, www.swissmedicinfo.ch,(última consulta: 06.02.2024)
- Mayorandan S, et al: Estudio transversal de 168 pacientes con tirosinemia hepatorrenal e implicaciones para la práctica clínica. Orphanet J Rare Dis 2014; 9: 107.
- McKiernan PJ, Preece MA, Chakrapani A: Resultado de los niños con tirosinemia hereditaria tras el cribado neonatal. Arch Dis Child 2015; 100(8): 738-741.
- Bartlett DC, et al: El tratamiento precoz con nitisinona reduce la necesidad de trasplante hepático en niños con tirosinemia tipo 1 y mejora la función renal postrasplante. J Inherit Metab Dis 2014; 37(5): 745-752.
- Önenli Mungan N, et al: Tirosinemia tipo 1 y crisis neurológica irreversible tras un mes de interrupción de la nitisona. Metab Brain Dis 2016; 31(5): 1181-1183.
- Schlump JU, et al: Crisis neurológica grave en un paciente con tirosinemia hereditaria tipo I tras la interrupción del tratamiento con NTBC. J Inherit Metab Dis 2008; 31 Suppl 2: S223-S225.
- Uçar HK, et al: Informe de un caso de una asociación muy rara de tirosinemia tipo I y pancreatitis que imita una crisis neurológica de tirosinemia tipo I. Balkan Med J 2016; 33(3): 370-372.
- Chinsky JM, et al: Diagnóstico y tratamiento de la tirosinemia tipo I: revisión y recomendaciones de un grupo de consenso estadounidense y canadiense. Genet Med 2017; 19(12): doi:10.1038/gim.2017.101
- de Laet C, et al: Recomendaciones para el tratamiento de la tirosinemia tipo 1. Orphanet J Rare Dis 2013; 8: 8. Publicado el 11 de enero de 2013. doi:10.1186/1750-1172-8-8
- Alvarez F, Mitchell GA: Tirosinemia y trasplante de hígado: experiencia en el CHU Sainte-Justine. Adv Exp Med Biol 2017; 959: 67-73.
- De Laet C, et al: Resultados neuropsicológicos de los pacientes con tirosinemia tipo 1 tratados con NTBC. Dev Med Child Neurol 2011; 53(10): 962-964.
- Morrow G, Angileri F, Tanguay RM: Aspectos moleculares de las mutaciones FAH implicadas en la enfermedad HT1. Adv Exp Med Biol 2017; 959: 25-48.
- Morrow G, Tanguay RM: Aspectos bioquímicos y clínicos de la tirosinemia hereditaria tipo 1 Adv Exp Med Biol 2017; 959: 9-21.
- van Spronsen FJ, et al: Consideraciones dietéticas en la tirosinemia tipo I. Adv Exp Med Biol 2017; 959: 197-204.
- Blackburn PR, et al: Tirosinemia silenciosa tipo I sin tirosina o succinilacetona elevadas asociada a cirrosis hepática y carcinoma hepatocelular. Hum Mutat 2016; 37(10): 1097-1105.
PRÁCTICA MÉDICA GENERAL 2024; 19(2): 36-37