
Además de los cuidados estándar para todas las formas de ELA, se han producido avances significativos, en particular para las formas monogenéticas de ELA, gracias al desarrollo de métodos terapéuticos específicos basados en genes, actualmente la ASO en particular. El diagnóstico genético básico, al menos de los genes más comunes (SOD1, C9orf72, FUS, TARDP), se recomienda por tanto a todos los pacientes con ELA en el momento del diagnóstico.
Puede realizar el examen CME en nuestra plataforma de aprendizaje después de revisar los materiales recomendados. Haga clic en el siguiente botón:
Los casos de esclerosis lateral amiotrófica (ELA) fueron descritos por primera vez por Jean Martin Charcot en 1873 [1]. Charcot ya aportó pruebas neuropatológicas de una degeneración subyacente del sistema motor. Ahora se sabe que la esclerosis lateral amiotrófica es una neurodegeneración multisistémica con numerosas manifestaciones extramotoras, a pesar de la degeneración predominante de las neuronas motoras primera y segunda y del tracto corticoespinal. Hemos adquirido cada vez más conocimientos sobre los factores genéticos subyacentes, sobre todo en la última década, lo que también ha dado lugar a las primeras consecuencias terapéuticas directas.
Epidemiología
Basándose en los datos del registro de pacientes mejor gestionado de Suabia, puede estimarse que en el conjunto de Alemania hay entre 8.000 y 9.000 enfermos de ELA [2]. Con una edad media de aparición de 70 a 75 años y un ligero predominio masculino, se supone una incidencia de aproximadamente 3/100.000 pacientes. La prevalencia a lo largo de la vida como medida estadística más descriptiva de la probabilidad de desarrollar ELA es de 1:400. Estas cifras epidemiológicas son muy congruentes con las de otros países europeos. En otras partes del mundo, como Asia, los datos epidemiológicos son diferentes [3,4].
Síntomas clínicos
Los síntomas centrales de la ELA son una paresia de inicio focal, progresiva y atrófica con frecuentes espasmos musculares y fasciculaciones como signo de un daño creciente de las segundas neuronas motoras a nivel espinal o en la zona del tronco encefálico [5]. Esto va precedido o acompañado de una afectación de las primeras motoneuronas en la corteza motora primaria y el tracto corticoespinal con aumento y salto de los reflejos de estiramiento muscular y evidencia de reflejos patológicos o un aumento del tono muscular en el sentido de la espasticidad.
Clínicamente y en la vida cotidiana se encuentran déficits cognitivos relevantes y anomalías conductuales en el sentido de un síndrome de demencia frontotemporal como síntomas extramotores en aproximadamente el 5% de las personas afectadas por ELA [6,7]. En los últimos años se han descrito cada vez más trastornos acompañantes del sistema nervioso autónomo [8]. Además, el dolor de diversos orígenes también es importante durante el curso de la enfermedad [9].
Procedimientos de diagnóstico y diagnóstico
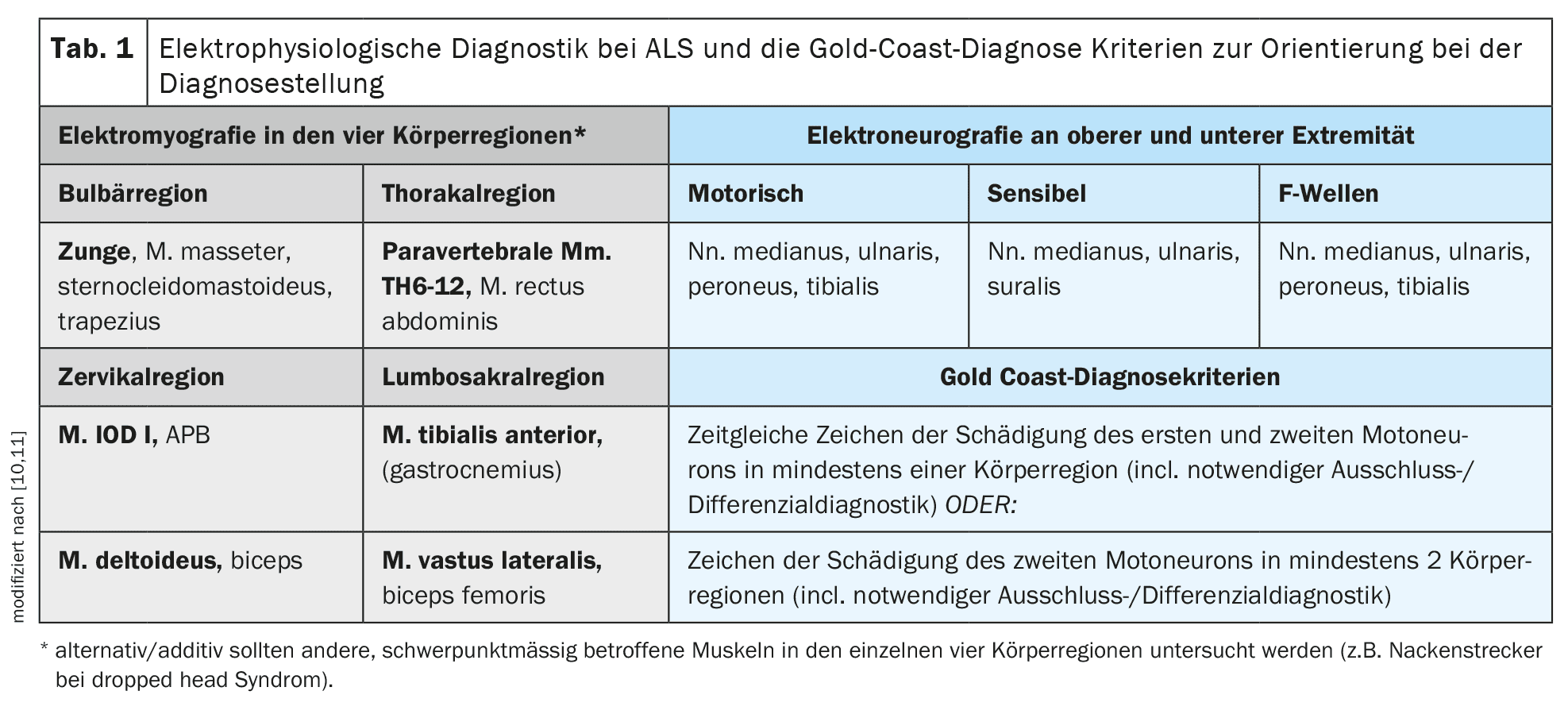
El diagnóstico se realiza principalmente de forma clínica tras la exclusión coherente y cuidadosa de diagnósticos diferenciales alternativos. [10]Los criterios de Gold Coast proporcionan una buena orientación para el diagnóstico clínico. El procedimiento diagnóstico de apoyo más importante es la electromiografía para detectar la denervación activa o los potenciales de fasciculación politópica con evidencia simultánea de daño neurogénico crónico. En la electromiografía deben examinarse diferentes músculos de las cuatro regiones corporales (nervios craneales/bulbares – cervicales – torácicos – lumbosacros) [11]. Además de la electromiografía, también puede realizarse una ecografía muscular para determinar la presencia de fasciculaciones musculares politópicas y evaluar el trofismo muscular y la estructura interna. La ecografía muscular puede ser especialmente ventajosa para la detección de fasciculaciones musculares en músculos más grandes o profundos, como partes del músculo cuádriceps femoral o los músculos de la base de la lengua, y puede ser un complemento importante de la electromiografía. La electroneurografía de los nervios motores y sensoriales, así como el diagnóstico por ondas F en las extremidades superiores e inferiores, son especialmente necesarios para excluir la neuropatía desmielinizante primaria y, en particular, los bloqueos de conducción (CIDP o neuropatía motora multifocal/MMN).
La estimulación magnética transcraneal para registrar los potenciales evocados motores está disponible para objetivar y, en caso necesario, cuantificar la afectación de las primeras neuronas motoras. Este método de examen puede utilizarse para examinar la conducción motora central de los axones espesamente mielinizados del tracto corticoespinal desde la corteza motora como lugar de estimulación a lo largo de toda la médula espinal [12]. Tiene sentido examinar un músculo distal de cada una de las extremidades superiores e inferiores. La tabla 1 ofrece una visión detallada de los diagnósticos electrofisiológicos propuestos para la ELA y los criterios diagnósticos de Gold Coast.
Las cadenas ligeras de neurofilamentos (NfL) son un biomarcador importante . Pueden determinarse fácilmente en suero y líquido cefalorraquídeo [13]. Es importante que la determinación se lleve a cabo utilizando un método de detección suficientemente sensible en un laboratorio establecido con valores límite ajustados a la edad. Debe subrayarse que, aunque la NfL puede representar un componente adicional importante para el diagnóstico, una NfL dentro del rango normal no puede descartar el diagnóstico de ELA y también pueden darse valores elevados de NfL en otros diagnósticos diferenciales de ELA (CIDP, neuropatía vasculítica, neuropatía asociada a amiloidosis, MMN) [14]. La importancia de la NfL como marcador pronóstico de progresión parece ser incluso mayor que en los diagnósticos diferenciales, aunque tampoco en este caso puede hacerse una afirmación fiable para el paciente individual basándose en el valor de NfL.
Aunque el diagnóstico genético seguía considerándose opcional en la directriz S1, vigente y disponible desde 2021, los autores creen que esta situación ha cambiado como consecuencia de los avances de los últimos tres años [5]. [15,16]A la vista de los avances terapéuticos en este campo, todos los pacientes con ELA deberían someterse obligatoriamente a pruebas genéticas específicas al menos para detectar la presencia de una mutación en el gen Cu/Zn de la superóxido dismutasa 1 (SOD 1) y los pacientes jóvenes con ELA menores de 40 años para detectar la presencia de una mutación patogénica en el gen FUS. Opcionalmente, también deberían realizarse diagnósticos genéticos más amplios con el examen de otros genes como el C9orf72, el TARDP, el TBK1, etc. [17]. Las siguientes secciones sobre etiología y genética y, en particular, sobre terapia tratarán estas cuestiones con más detalle. La tabla 1 ofrece una visión general de los procedimientos diagnósticos descritos y de los criterios diagnósticos de Gold Coast recomendados como guía en la práctica clínica.
Con respecto a otras investigaciones clínicamente relevantes para realizar diagnósticos diferenciales y de exclusión exhaustivos en función de la presentación clínica inicial (por ejemplo, imágenes por resonancia magnética, diagnósticos ORL, diagnósticos de laboratorio), así como diagnósticos relevantes durante el curso de la enfermedad para evaluar el pronóstico (por ejemplo, prueba de función pulmonar/prueba de función diafragmática, diagnóstico de la deglución mediante FEES, psicometría mediante ECAS), nos gustaría remitirnos explícitamente a la presentación tan detallada y clara de la directriz S1 [5].
Espectro fenotípico y patrón de la paresia muscular en el contexto de la neuroanatomía y la fisiopatología de la ELA
Los descubrimientos científicos de las dos últimas décadas, en particular los estudios neuroanatómicos y neuropatológicos, han cambiado fundamentalmente la visión de la ELA. Hoy en día, la ELA ya no se considera una degeneración puramente del sistema motor, sino una degeneración multisistémica [18,19].
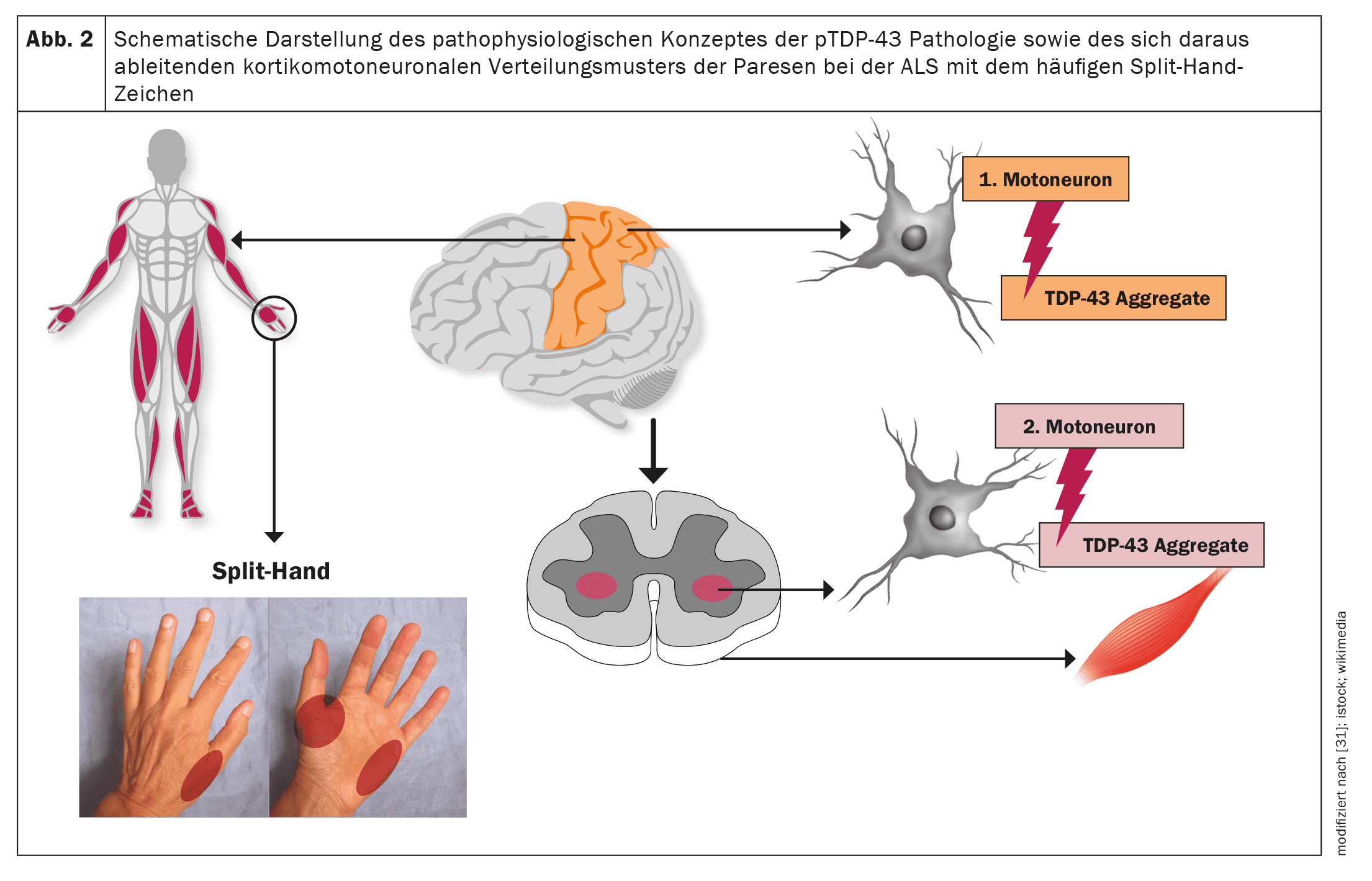
La ELA, al igual que otras enfermedades neurodegenerativas como el Alzheimer o la enfermedad de Parkinson, es una proteinopatía, es decir, los agregados proteicos patológicos en las células nerviosas motoras como correlato neuropatológico conducen a la disfunción de las células nerviosas motoras afectadas y, en última instancia, a la apoptosis y, por tanto, a la pérdida de células nerviosas motoras. En el caso de la ELA, en más del 95% de los casos se trata de agregados de la proteína TDP-43 [20]. Sólo en el caso de variantespatológicas subyacentes de SOD1 o FUSse produce una neurodegeneración independiente de TDP-43 [21]. Los estudios neuropatológicos han establecido una propagación cerebral escalonada y por etapas de la patología del TDP-43 en la ELA, comparable a la patología de la α-sinucleína en la enfermedad de Parkinson y a la patología de la tau en la enfermedad de Alzheimer [22]. Estos hallazgos, así como el patrón corticomotoneuronal de la paresia con el signo de la mano partida (atrofia asimétrica de los músculos C8/T1 o [23–26] músculos de la mano provistos del cúbito) como signo clínico muy común en la ELA implican un origen de los cambios neuropatológicos en la zona de la corteza motora primaria y una propagación gradual de la patología TDP-43 desde allí mediante transporte axonal a las segundas neuronas motoras, es decir, los núcleos motores de los nervios craneales en el tronco encefálico y las células del asta anterior de la médula espinal.
Parece producirse una propagación de los cambios neuropatológicos similar a la de los priones, lo que explicaría el inicio focal de la manifestación motora, así como una propagación gradual a miotomas y regiones corporales vecinas [27,28]. En consecuencia, dependiendo de la manifestación inicial de la neuropatología, el espectro fenotípico de la ELA -con respecto a los síntomas motores únicamente- no es uniforme, sino muy variable [29]. Por ejemplo, pueden darse fenotipos motores topográficamente distintos. Además, el fenotipo está influido por la velocidad de degeneración, normalmente diferente, de la primera y la segunda neuronas motoras y por los síntomas clínicos asociados. Sin que hasta la fecha se conozcan con precisión los mecanismos moleculares, en este caso parece ser decisiva la relación entre los agregados insolubles de proteína TDP-43, que ya no pueden transportarse axonalmente y, por tanto, se acumulan localmente, y los oligómeros solubles de TDP-43 como precursores de los agregados de TDP-43. Evidentemente, estos precursores solubles se transportan entonces por vía axonal desde las primeras motoneuronas hasta las segundas motoneuronas, donde se depositan entonces obligatoriamente como agregados proteicos y conducen a la muerte de estas células [23].
Hasta la fecha, no existe una clasificación estandarizada de los fenotipos clínicos que tenga en cuenta la proporción o el foco del daño en la primera y la segunda neurona motora, el patrón clínico o el foco de la paresia atrófica y su propagación.
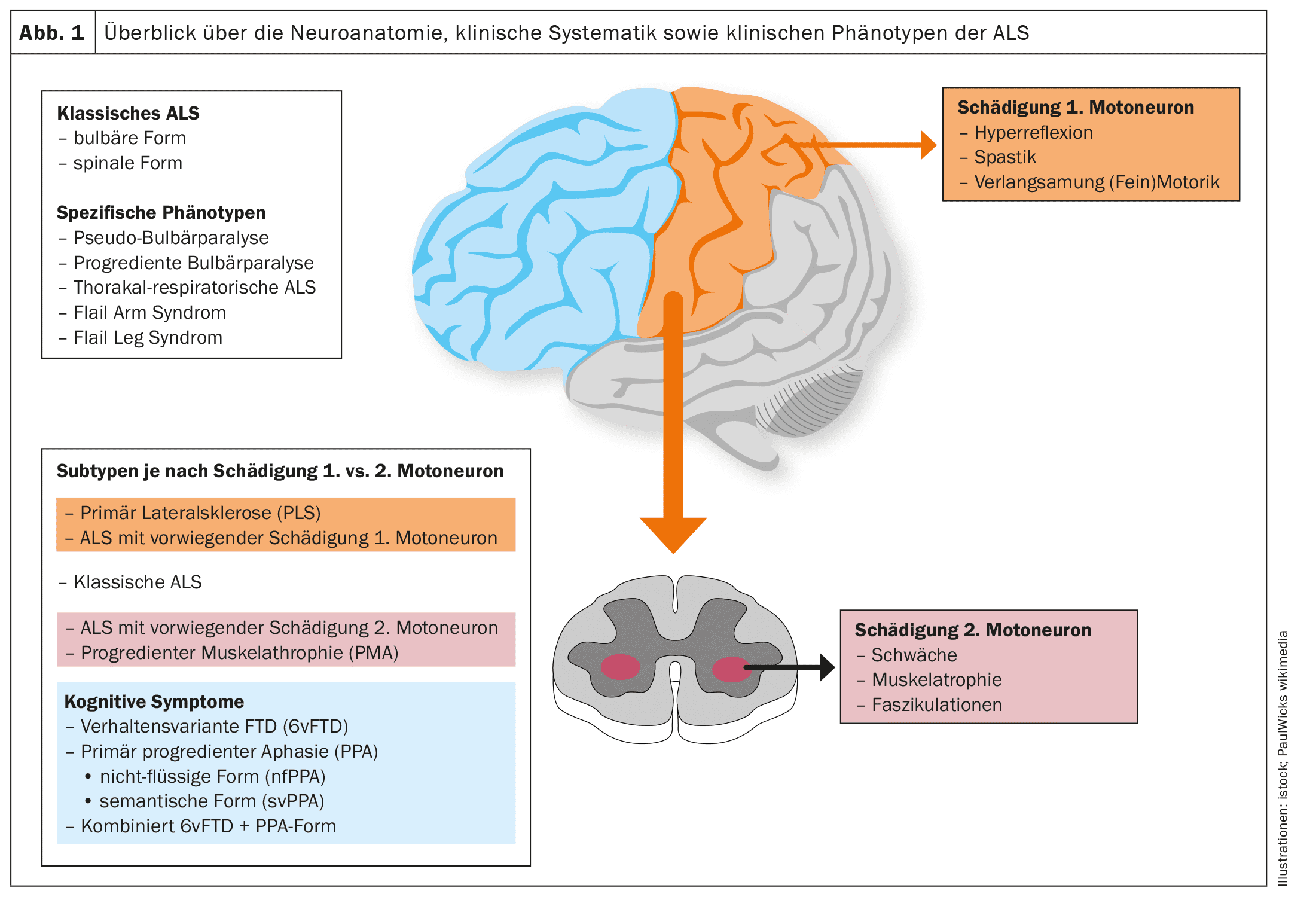
Las figuras 1 y 2 ofrecen una visión esquemática de la neuroanatomía y la fisiopatología de la enfermedad, así como de los fenotipos clínicos y su clasificación.
Etiología y genética
La etiología de la ELA aún no se conoce suficientemente bien. En la forma esporádica de la enfermedad, que representa la gran mayoría de los casos, se supone una génesis multifactorial con la interacción de diversos factores ambientales externos desfavorables, factores metabólicos epigenéticos y una posible susceptibilidad genética adicional. Los cambios en el transporte nuclear-citosólico de proteínas, el metabolismo del ARN, la función oxidativa celular mitocondrial, la excitabilidad glutamatérgica, el transporte axonal de proteínas y la autofagia celular se discuten como los mecanismos metabólicos intracelulares subyacentes, mientras que los trastornos funcionales de los atrocitos y los oligodendrocitos, así como los procesos inflamatorios, se discuten como mecanismos extracelulares aditivos [31]. Se han obtenido conocimientos significativos sobre los posibles mecanismos etiológicos de la ELA esporádica principalmente a través de formas genéticas con procesos fisiopatológicos distintos.
[32]En 1993, se describieron por primera vez las mutaciones patogénicas en la superóxido dismutasa 1 Cu/Zn (SOD1) como causa genética de la ELA . En la actualidad se conoce un gran número de mutaciones genéticas que constituyen la etiología subyacente de la ELA. En el caso de la ELA aparentemente esporádica sin antecedentes familiares llamativos en Alemania, puede detectarse una causa monogenética en algo más del 10% de los casos [17]. Las causas genéticas más comunes en Alemania son las expansiones patológicas de la repetición C9orf72 y las mutaciones en los genes SOD1, TARDP y FUS [17].Por lo tanto, el diagnóstico genético en el momento del diagnóstico debería ofrecerse a todos los pacientes con ELA, ya que puede tener consecuencias terapéuticas inmediatas, de las que hablaremos en detalle en la siguiente sección.
Terapia y pronóstico
El riluzol está aprobado en Alemania desde 1996 como la única sustancia farmacológica con un efecto positivo demostrado en la progresión de la ELA. Se postula como mecanismo de acción decisivo una reducción de la liberación de glutamato y, por tanto, de la excitotoxicidad. [18]Esta hipótesis se ha visto reforzada por la evidencia de una degeneración primaria de las vías glutamatérgicas corticofugales . Una dosis diaria de riluzol de 100 mg presenta el mejor perfil de eficacia/efectos secundarios. Los análisis retrospectivos de un total de diez registros clínicos de ELA aportan pruebas de una prolongación media del tiempo de supervivencia de hasta 19 meses con riluzol, y también hay indicios claros de que esta sustancia también es eficaz en estadios más avanzados de la enfermedad [33–35]. En general, el riluzol se tolera bien; entre los efectos secundarios conocidos se incluyen un aumento de las transaminasas, que por lo tanto deben controlarse regularmente, y molestias gastrointestinales. En la actualidad también se dispone de formas de dosificación alternativas, como zumos o comprimidos bucodispersables, para los pacientes de ELA con disfagia.
Además de la terapia farmacológica con riluzol, se considera que la prevención de un estado metabólico catabólico con una pérdida de peso consecutiva tiene un beneficio pronóstico adicional. Cuanto mayor sea el índice de masa corporal (IMC), más favorable será el pronóstico [36].
Las recomendaciones actuales para los pacientes con ELA son, por tanto, mantener un peso estable y evitar la pérdida de peso. En este contexto, la inserción de una sonda PEG también desempeña un papel importante en la disfagia progresiva, con un tiempo de supervivencia prolongado como resultado de esta medida [37]. En la actualidad se está investigando intensamente en estudios si los enfoques terapéuticos anticatabólicos específicos, como las intervenciones nutricionales específicas altas en grasas y calorías o las intervenciones nutricionales cetogénicas, pueden mejorar el pronóstico y en qué medida. En este contexto, es de gran importancia el estudio LIPCAL-ALS 2 en toda Alemania comoensayo iniciado por el investigador (IIT) realizado por colegas de Ulm, cuyo inicio está previsto entre 2024 y 2025.
La ventilación no invasiva (VNI) e invasiva mediante un traqueostoma es otra medida importante para prolongar el tiempo de supervivencia en la ELA con insuficiencia ventilatoria [38,39]. Esto también es plausible, ya que la hipoventilación alveolar con hipercapnia consecutiva debida a la afectación diafragmática suele ser un factor importante en la muerte de los pacientes con ELA después de tres a cinco años de progresión de la enfermedad.
El desarrollo de oligonucleótidos antisentido (ASO) para formas específicas de ELA inducidas genéticamente puede considerarse un hito. En particular, cabe mencionar aquí el ASO Tofersen, administrado por vía intratecal, que se une al ARNm de la SOD1en pacientes con variantespatogénicas de la SOD1 (alrededor del 1–2% de todos los casos de ELA) e impide así la expresión citotóxica de la proteína SOD1. [16,40]Se ha demostrado que el efecto mecánico de una reducción significativa de la expresión de SOD1 en aproximadamente un 30% en humanos se produce muy rápidamente a los pocos días de iniciar la terapia con Tofersen, seguido de un descenso significativo de la NfL en el LCR y el suero, antes de que finalmente se produzca una ralentización del descenso de la puntuación ALSFRS-R . Tras una observación a más largo plazo de doce meses, también se observaron otras señales positivas con relevancia clínica, como efectos sobre la función muscular y la estabilización del peso.
En EE.UU., Tofersen fue aprobado por la FDA en abril de 2023 únicamente sobre la base de los convincentes datos de biomarcadores con un descenso significativo de los valores de NfL. En Alemania, Tofersen se puso a disposición como parte de un programa de acceso abierto. Los datos iniciales de aplicación en el mundo real de la red neuronal motora alemana confirmaron de forma impresionante los datos del estudio de Tofersen con datos aún mejores en términos de parámetros de progresión clínica [41]. En este contexto, era lógico que la EMA se pronunciara a favor de la aprobación de Tofersen en febrero de 2024.
Además del ASO Tofersen para la ELA asociada a SOD1, actualmente se están desarrollando ASO o ya se han investigado en estudios, en particular contra C9orf72 y FUS [15,42]. En particular, cabe mencionar aquí el ASO Jacifusen (ION363) cuando se detecta unavariante patógena del FUS como causa de la ELA juvenil, que se está probando actualmente en un estudio multicéntrico y multinacional con dos centros de estudio (Rostock y Ulm) en Alemania.
La escala revisada de valoración funcional de la ELA (ALSFRS-R) es una puntuación probada y bien establecida para evaluar las funciones motoras de las cuatro regiones corporales y no sólo es un criterio de valoración importante para los estudios clínicos, sino que también ha demostrado ser un parámetro de seguimiento fácilmente factible en la práctica clínica [43]. Se espera que la ALSFRS-SE (SE: “autoexplicativa”), que se ha acordado recientemente en la red alemana de EMN en alemán con las correspondientes explicaciones concretas y ejemplares para los ítems individuales y las deficiencias funcionales, tenga una ventaja adicional en términos de manejo práctico y, en particular, de precisión diagnóstica [44].
Conclusión para la práctica
Además de los cuidados estándar para todas las formas de ELA (riluzol, prevención del estado metabólico catabólico, incluida la colocación a tiempo de una sonda PEG, ventilación precoz no invasiva y, si es necesario, invasiva en caso de insuficiencia ventilatoria), se han producido avances significativos, en particular para las formas monogenéticas de ELA, gracias al desarrollo de métodos terapéuticos específicos basados en genes, actualmente ASO en particular. Cabe mencionar aquí el Tofersen como terapia específica muy eficaz para la ELA asociada a SOD1, que ahora también está disponible en Alemania.
Por lo tanto, se recomienda un diagnóstico genético básico, al menos de los genes más comunes (SOD1, C9orf72, FUS, TARDP), para todos los pacientes con ELA en el momento del diagnóstico. Es de esperar que los próximos estudios en este campo demuestren hasta qué punto las intervenciones específicas, anticatabólicas y de alto contenido calórico pueden influir favorablemente en la evolución de la ELA esporádica.
Mensajes para llevar a casa
- El riluzol 100 mg/día, la prevención de un estado metabólico catabólico, incluida la colocación oportuna de una sonda PEG y la ventilación no invasiva precoz en caso de insuficiencia respiratoria, están disponibles como cuidados estándar para todas las formas de ELA. colocación oportuna de una sonda PEG, así como la ventilación temprana no invasiva y, en caso necesario, invasiva en caso de insuficiencia respiratoria.
- Además, se han logrado avances significativos, en particular para las formas monogenéticas de ELA. La base de ello es el desarrollo de métodos terapéuticos específicos basados en genes, como los oligonucleótidos antisentido (ASO) en particular. Cabe mencionar aquí el Tofersen como una terapia muy eficaz y específica para la ELA asociada a SOD1.
- Por lo tanto, se recomienda un diagnóstico genético básico, al menos de los genes más comunes (SOD1, C9orf72, FUS, TARDP) para todos los pacientes con ELA en el momento del diagnóstico.
Literatura:
- Duyckaerts C, Maisonobe T, Hauw JJ, Seilhean D: Charcot identifica e ilustra la esclerosis lateral amiotrófica. Neuropatología libre. 2. doi:10.17879/freeneuropathology-2021-3323.
- Uenal H, Rosenbohm A, Kufeldt J, et al: Incidencia y variación geográfica de la esclerosis lateral amiotrófica (ELA) en el sur de Alemania: exhaustividad del registro de ELA de Suabia. PLoS ONE. 2014; 9(4). doi:10.1371/journal.pone.0093932.
- Jun KY, Park J, Oh KW, et al: Epidemiología de la ELA en Corea mediante big data a escala nacional. J Neurol Neurosurg Psychiatry. 2019;90(4): 395-403. doi:10.1136/jnnp-2018-318974.
- Marin B, Boumédiene F, Logroscino G, et al: Variación de la incidencia mundial de la esclerosis lateral amiotrófica: un metaanálisis. Int J Epidemiol 2017; 46(1): 57-74. doi:10.1093/ije/dyw061.
- Petri SA-O GJ Ludolph AC. “Enfermedades de la neurona motora” de la Sociedad Alemana de Neurología (DGN). (2524-2348; Electrónico).
- Finsel J, Uttner I, Vázquez Medrano CR, et al: Cognición en el curso de la ELA: un metaanálisis. Amyotroph Lateral Scler Front Degener. 2023; 24(1-2): 2-13. doi:10.1080/21678421.2022.2101379.
- Iazzolino B, Pain D, Peotta L, et al: Validación de la clasificación revisada del deterioro cognitivo y conductual en la ELA. J Neurol Neurosurg Psychiatry 2019; 90(7): 734-739. doi:10.1136/jnnp-2018-319696.
- Oprisan AL, Popescu BO: Disautonomía en la esclerosis lateral amiotrófica. Int J Mol Sci 2023; 24(19). doi:10.3390/ijms241914927.
- Chiò A, Mora G, Lauria G. Dolor en la esclerosis lateral amiotrófica. Lancet Neurol 2017; 16(2): 144-157. doi:10.1016/S1474-4422(16)30358-1
- Shefner JM, Al-Chalabi A, Baker MR, et al: Una propuesta de nuevos criterios diagnósticos para la ELA. Clin Neurophysiol 2020; 131(8): 1975-1978. doi:10.1016/j.clinph.2020.04.005.
- Koch JC, Petri S, Zeller D: Diagnóstico electrofisiológico de la sospecha de esclerosis lateral amiotrófica – Recomendaciones de consenso de la Red alemana de enfermedades de las neuronas motoras (MND-NET). Clin Neurophysiol 2024. 2024; 55: 82-88.
- Zoccolella S, Mastronardi A, Scarafino A, et al: Potenciales evocados motores en la esclerosis lateral amiotrófica: implicaciones potenciales en la detección de la afectación subclínica de la UMN en el fenotipo de neurona motora inferior. J Neurol 2020; 267(12): 3689-3695. doi:10.1007/s00415-020-10073-5.
- Shahim P, Norato G, Sinaii N, et al. Neurofilamentos en la esclerosis lateral amiotrófica esporádica y familiar: una revisión sistemática y metaanálisis. Genes. 2024;15(4). doi:10.3390/genes15040496.
- Behzadi A, Pujol-Calderón F, Tjust AE, et al: Los neurofilamentos pueden diferenciar los subgrupos de ELA y la ELA de los imitadores comunes de diagnóstico. Sci Rep 2021; 11. doi:10.1038/s41598-021-01499-6.
- Korobeynikov VA, Lyashchenko AK, Blanco-Redondo B, et al: Silenciamiento con oligonucleótidos antisentido de la expresión de FUS como enfoque terapéutico en la esclerosis lateral amiotrófica. Nat Med 2022; 28(1): 104-116. doi:10.1038/s41591-021-01615-z.
- Miller TM, Cudkowicz ME, Genge A, et al: Ensayo del oligonucleótido antisentido Tofersen para la ELA SOD1. N Engl J Med. Publicado en línea el 22 de septiembre de 2022. doi:10.1056/NEJMoa2204705.
- Ruf WP, Boros M, Freischmidt A, et al: Espectro y frecuencia de las variantes genéticas en la esclerosis lateral amiotrófica esporádica. Brain Commun 2023; 5(3). doi:10.1093/braincomms/fcad152.
- Braak H, Brettschneider J, Ludolph AC, et al: Esclerosis lateral amiotrófica: un modelo de propagación axonal corticofugal. Nat Rev Neurol 2013; 9(12): 708-714. doi:10.1038/nrneurol.2013.221.
- Brettschneider J, Del Tredici K, Toledo JB, et al: Estadios de la patología pTDP-43 en la esclerosis lateral amiotrófica. Ann Neurol 2013; 74(1): 20-38. doi:10.1002/ana.23937.
- Neumann M: Neuropatología molecular de las proteinopatías TDP-43. Int J Mol Sci 2009; 10(1): 232-246. doi:10.3390/ijms10010232.
- Saberi S, Stauffer JE, Schulte DJ, Ravits J: “Neuropatología de la esclerosis lateral amiotrófica y sus variantes”. Neurol Clin 2015; 33(4): 855-876. doi:10.1016/j.ncl.2015.07.012.
- Braak H, Braak E: Estadificación de los cambios neurofibrilares relacionados con la enfermedad de Alzheimer. Neurobiol Aging 1995; 16(3): 271-278-284.
- Braak H, Ludolph AC: Neumann M, Ravits J, Del Tredici K. Los cambios patológicos del TDP-43 en las células de Betz difieren de los de las α-motoneuronas bulbares y espinales en la esclerosis lateral amiotrófica esporádica.
Acta Neuropathol (Berl) 2017; 133(1): 79-90. doi:10.1007/s00401-016-1633-2. - Eisen A, Braak H, Del Tredici K, et al: Las influencias corticales impulsan la esclerosis lateral amiotrófica. J Neurol Neurosurg Psychiatry 2017; 88(11): 917-924. doi:10.1136/jnnp-2017-315573.
- Ludolph AC, Emilian S, Dreyhaupt J, et al: El patrón de paresia en la ELA es coherente con la fisiología de las proyecciones corticomotoneuronales a diferentes grupos musculares. J Neurol Neurosurg Psychiatry 2020; 91(9): 991-998. doi:10.1136/jnnp-2020-323331.
- Menon P, Kiernan MC, Vucic S: La hiperexcitabilidad cortical precede a la disfunción de la neurona motora inferior en la ELA. Clin Neurophysiol 2015; 126(4): 803-809. doi:10.1016/j.clinph.2014.04.023.
- Prasad A, Bharathi V, Sivalingam V, et al: Mecanismos moleculares del mal plegamiento del TDP-43 y patología en la esclerosis lateral amiotrófica. Front Mol Neurosci 2019;12. doi:10.3389/fnmol.2019.00025.
- Gosset P, Camu W, Raoul C, Mezghrani A: Prionoides en la esclerosis lateral amiotrófica. Brain Commun 2022;4(3). doi:10.1093/braincomms/fcac145.
- Hardiman O, Al-Chalabi A, Chio A, et al: Esclerosis lateral amiotrófica. Nat Rev Dis Primer 2017; 3:17071. doi:10.1038/nrdp.2017.71.
- Masrori P, Van Damme P: Esclerosis lateral amiotrófica: una revisión clínica. Eur J Neurol 2020; 27(10): 1918-1929. doi:10.1111/ene.14393.
- Eisen A, Vucic S, Mitsumoto H: Historia de la ELA y las teorías contrapuestas sobre su patogenia: capítulo del manual de la IFCN. Clin Neurophysiol Pract 2023; 9: 1-12. doi:10.1016/j.cnp.2023.11.004.
- Rosen DR, Siddique T, Patterson D, et al: Las mutaciones en el gen de la superóxido dismutasa Cu/Zn están asociadas a la esclerosis lateral amiotrófica familiar. Nature 1993; 362(6415): 59-62. doi:10.1038/362059a0.
- Bensimon G, Lacomblez L, Meininger V, Grupo el AS: Un ensayo controlado de riluzol en la esclerosis lateral amiotrófica. http://dx.doi.org/10.1056/NEJM199403033300901.
- Hinchcliffe M, Smith A: Riluzol: las pruebas en el mundo real apoyan una ampliación significativa de los tiempos medios de supervivencia en pacientes con esclerosis lateral amiotrófica. Degener Neurol Neuromuscul Dis 2017;7: 61-70. doi:10.2147/DNND.S135748.
- Miller RG, Mitchell JD, Moore DH: Riluzol para la esclerosis lateral amiotrófica (ELA)/enfermedad de la motoneurona (EMN). Cochrane Database Syst Rev 2012; 2012(3). doi:10.1002/14651858.CD001447.pub3.
- Dupuis L, Pradat PF, Ludolph AC, Loeffler JP: Metabolismo energético en la esclerosis lateral amiotrófica. Lancet Neurol 2011; 10(1): 75-82. doi:10.1016/S1474-4422(10)70224-6.
- Cui F, Sun L, Xiong J, et al: Efectos terapéuticos de la gastrostomía endoscópica percutánea sobre la supervivencia en pacientes con esclerosis lateral amiotrófica: Un metaanálisis. PLoS ONE 2018; 13(2). doi:10.1371/journal.pone.0192243.
- Dorst J, Ludolph AC: Ventilación no invasiva en la esclerosis lateral amiotrófica. Ther Adv Neurol Disord 2019; 12: 1756286419857040. doi:10.1177/1756286419857040.
- Radunovic A, Annane D, Rafiq MK, et al: Ventilación mecánica para la esclerosis lateral amiotrófica/enfermedad de la motoneurona.
Cochrane Database Syst Rev 2017; 2017(10). doi:10.1002/14651858.CD004427.pub4. - Miller T, Cudkowicz M, Shaw PJ, et al: Ensayo de fase 1-2 del oligonucleótido antisentido Tofersen para la ELA SOD1. N Engl J Med 2020; 383(2): 109-119. doi:10.1056/NEJMoa2003715.
- Wiesenfarth M, Dorst J, Brenner D, et al: Efectos del tratamiento con tofersen en pacientes con SOD1-ALS en un entorno “real” – un estudio de cohortes multicéntrico de 12 meses del programa alemán de acceso temprano. eClinicalMedicine. 2024; 69. doi:10.1016/j.eclinm.2024.102495.
- Meijboom KE, Brown RH: Aproximaciones a la terapia de modulación génica para la ELA. Neuroterapéutica 2022;19(4): 1159-1179. doi:10.1007/s13311-022-01285-w.
- Cedarbaum JM, Stambler N, Malta E, et al: The ALSFRS-R: a revised ALS functional rating scale that incorporates assessments of respiratory function. Grupo de estudio de la ELA del BDNF (fase III). J Neurol Sci 1999; 169(1-2): 13-21. doi:10.1016/s0022-510x(99)00210-5.
- Maier A, Boentert M, Reilich P, et al: ALSFRS-R-SE: una versión adaptada, anotada y autoexplicativa de la escala revisada de valoración funcional de la esclerosis lateral amiotrófica. Neurol Res Pract 2022; 4. doi:10.1186/s42466-022-00224-6.
| Publicado por primera vez en neuro aktuell 2024; 38(8): 30-35. |
HAUSARZT PRAXIS 2024; 19(11): 12–17









