La esperanza y la calidad de vida de los pacientes con ELA pueden mejorar con una terapia moderna. La voluntad del paciente es primordial y debe determinarse siempre de nuevo. No hay novedades en la terapia farmacológica: el riluzol debe iniciarse pronto. Al principio de la enfermedad, es aconsejable una evaluación detallada en el entorno hospitalario de una clínica neurológica. Se recomienda seguir el tratamiento en centros especializados durante el curso de la enfermedad.
En la esclerosis lateral amiotrófica (ELA), la enfermedad motoneuronal más común, se produce una pérdida progresiva de células nerviosas en el sistema motor. Esto suele afectar tanto a las primeras motoneuronas del tracto piramidal como a las segundas motoneuronas de las células del asta anterior. Al mismo tiempo, la gravedad puede variar, sobre todo al principio de la enfermedad, y pueden predominar los signos de la primera o la segunda motoneurona. El espectro de las enfermedades de las neuronas motoras incluye otras enfermedades como la esclerosis lateral primaria (ELP) o la atrofia muscular espinal (AME), que a su vez sólo afectan al primer o segundo miembro. segunda neurona motora.
La incidencia de la ELA es rara en comparación con otras enfermedades. Sin embargo, la incidencia sigue siendo de alrededor de 2/100.000 habitantes, por lo que es sólo ligeramente inferior a la de la esclerosis múltiple, por ejemplo. En cambio, la prevalencia es muy baja, entre 3 y 8/100.000 habitantes [1]. Esto refleja indirectamente la corta supervivencia media de los pacientes tras el diagnóstico, que en la mayoría de los casos es de sólo dos a cuatro años. Curiosamente, sin embargo, alrededor del 10% de los pacientes tienen un curso mucho más lento con una supervivencia superior a los diez años.
En la actualidad, la terapia de apoyo moderna y máxima puede prolongar significativamente la supervivencia de los pacientes y mejorar su calidad de vida, al menos en lo que respecta a síntomas esenciales como el dolor, el hambre y la falta de aliento. Sin embargo, especialmente a la luz de las mejores posibilidades médicas, es muy importante situar siempre los deseos de los afectados en el centro de las decisiones terapéuticas [2]. Para ello, debe redactarse un testamento vital detallado en una fase temprana, que deberá revisarse una y otra vez a medida que avance la enfermedad.
Principios de diagnóstico
Las piedras angulares del diagnóstico en la ELA siguen siendo la elaboración de la historia clínica y el examen clínico. Si las lleva a cabo un neurólogo experimentado, normalmente ya hay pruebas claras de la presencia de un daño progresivo en la primera y/o segunda neurona motora. Esto por sí solo puede utilizarse para diagnosticar una ELA probable o segura según los criterios diagnósticos válidos [3]. Los hallazgos clínicos pueden corroborarse además mediante hallazgos electrofisiológicos, que ahora también se incluyen cada vez más en el algoritmo diagnóstico (Fig. 1) [4].
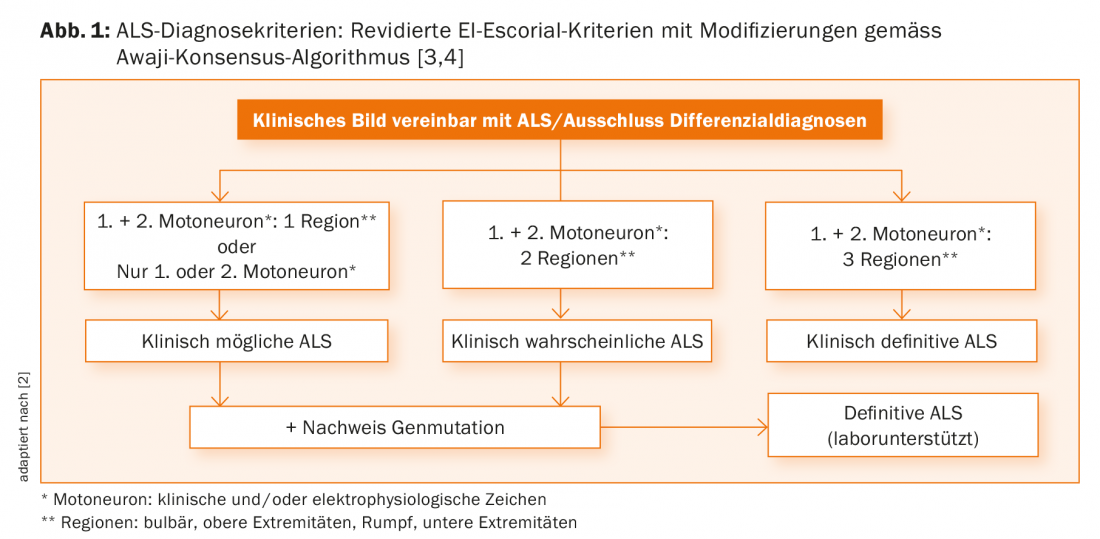
Desgraciadamente, con los criterios actuales, a menudo sólo se puede hacer un diagnóstico fiable en una fase tardía del curso de la enfermedad, lo que, por un lado, puede inquietar a los médicos remitentes, los pacientes y los familiares y, por otro, no proporciona una buena base para los estudios científicos. Por lo tanto, la revisión de los criterios diagnósticos sigue siendo de gran importancia.
Una tarea diagnóstica esencial es excluir cualquier diagnóstico diferencial que pueda causar una constelación comparable de síntomas. Éstas se resumen claramente en las directrices actuales de la EFNS [2]. A modo de ejemplo, la no tan rara combinación de estenosis espinal cervical y polineuropatía bien puede proporcionar la combinación de signos de primera y segunda neurona motora necesaria para el diagnóstico de ELA. Una evaluación hospitalaria en una clínica neurológica al inicio de la enfermedad ha demostrado ser eficaz para proporcionar espacio y tiempo suficientes para la evaluación clínica, la exclusión de diagnósticos diferenciales y la comunicación empática del diagnóstico.
Examen clínico
El examen clínico sirve en primer lugar para buscar signos de daños en la primera y segunda neurona motora en las cuatro regiones corporales (bulbar, extremidades superiores, tronco, extremidades inferiores). Los signos de la primera motoneurona incluyen espasticidad, clonía, hiperreflexia y fenómenos de desinhibición central. Los signos clínicos de la segunda motoneurona son principalmente fasciculaciones y atrofias. Además, deben buscarse signos atípicos en el examen clínico.
Por ejemplo, una afectación de los músculos oculares o también una falta de progresión dirigen la atención hacia diagnósticos alternativos. Por otra parte, las alteraciones sensoriales no descartan por completo la ELA, pero sin duda también deben ser motivo de una aclaración intensiva de los diagnósticos diferenciales.
Electrofisiología
La electroneurografía (ENG) puede utilizarse para descartar una polineuropatía. En la electromiografía (EMG), es típica de la ELA una constelación de síntomas consistente en signos de daño agudos, subagudos y posiblemente crónicos, que reflejan el curso de la enfermedad y la capacidad preservada de regeneración de los axones periféricos. Además de la búsqueda de actividad espontánea patológica en el músculo en reposo, se presta especial atención a la estabilidad y el tamaño de las unidades motoras en el análisis del potencial único. Un hallazgo patológico coincidente en el EMG es una parte esencial del diagnóstico de la ELA y puede considerarse igual a un signo clínico según los criterios de diagnóstico modificados [4].
Los potenciales evocados motores pueden detectar daños subclínicos en el tracto piramidal. Los métodos de cuantificación de las unidades motoras (estimación del número de unidades motoras) pueden describirse como métodos más bien científicos. Estos métodos podrían ser cada vez más importantes, especialmente para la progresión de la enfermedad y, por tanto, también para los ensayos clínicos [5].
Imágenes
La importancia de los ultrasonidos en el diagnóstico de la ELA ha ido en aumento en los últimos años, aunque no se espera que el método alcance la trascendencia que tiene en las enfermedades de los nervios periféricos. La ecografía nerviosa puede ser especialmente útil para descartar diagnósticos diferenciales relevantes como las neuropatías inmunomediadas. En concreto, los ultrasonidos también pueden utilizarse para examinar los músculos. Para la detección de fasciculaciones, la ecografía muscular, con su excelente sensibilidad y su especificidad algo menor, podría sustituir en el futuro, al menos parcialmente, a la electromiografía, sobre todo para los exámenes de seguimiento [6]. La resonancia magnética es especialmente importante para descartar diagnósticos diferenciales. En la mayoría de los casos, se visualizan el cerebro y toda la columna vertebral. Además, también se pueden obtener imágenes de los músculos y los nervios periféricos, aunque en la actualidad su interés se centra más en el ámbito científico que en la práctica clínica.
mentiras.
Diagnóstico genético
El diagnóstico genético en la ELA se ha ampliado en varios aspectos en los últimos años. Durante muchos años se han buscado, en particular, mutaciones en el gen de la superóxido dismutasa Cu/Zn SOD1, responsable de alrededor del 10-15% de los casos de ELA familiar. Paradójicamente, hay familias en las que la detección de la mutación no se correlaciona con la aparición de la enfermedad, lo que es un indicio de que deben existir otros factores o genes causantes de la enfermedad [7]. Entretanto, se ha encontrado un nuevo locus genético esencial a este respecto con el gen C9orf72. Las mutaciones en este gen son responsables de alrededor del 25% de los casos familiares y de alrededor del 10% de los casos esporádicos. Curiosamente, también existen mutaciones en este gen en una proporción similar de casos de demencia frontotemporal, lo que pone de relieve el vínculo entre ambas enfermedades también a nivel genético [8].
Las pruebas genéticas de las mutaciones más comunes están disponibles comercialmente en la actualidad. Por lo tanto, hay que insistir aún más en que el diagnóstico genético debe estar en manos experimentadas, especialmente en el caso de la ELA. Sólo debe solicitarse tras un examen y asesoramiento detallados por parte de neurólogos y genetistas humanos familiarizados con la enfermedad, ya que el resultado puede tener gran relevancia, especialmente para asesorar a familiares asintomáticos.
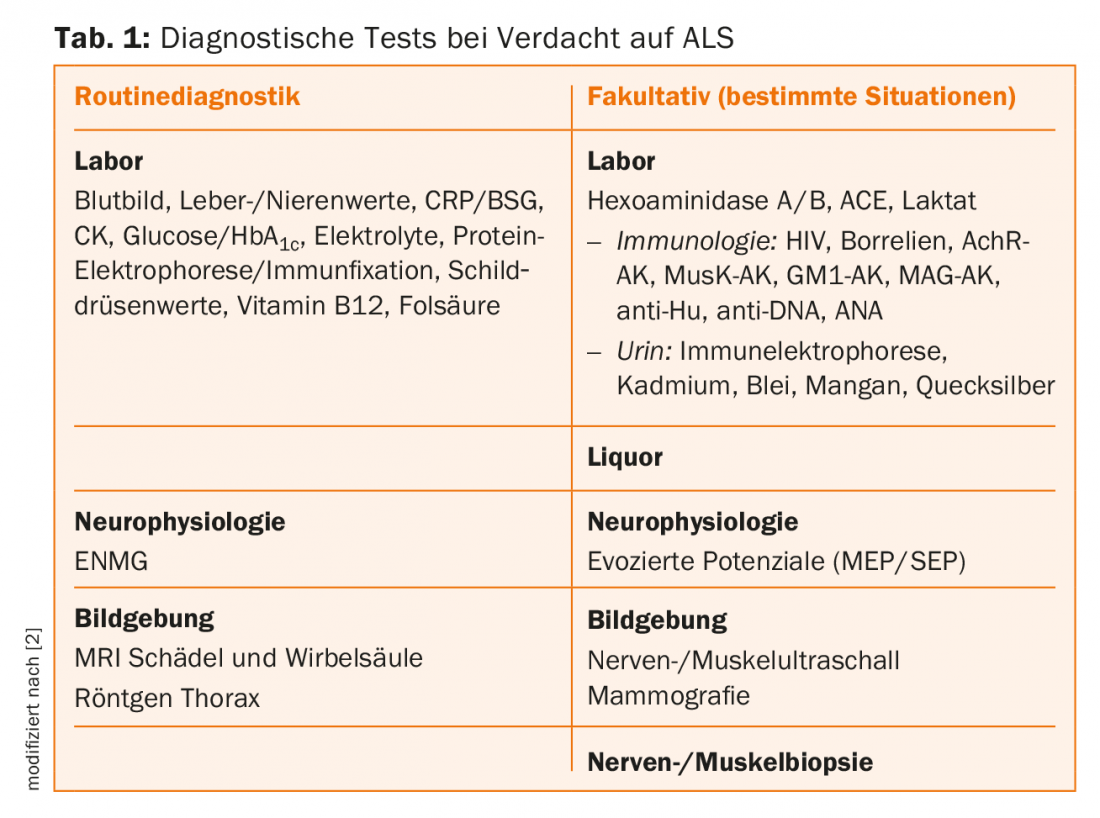
Otros diagnósticos
Si se sospechan anomalías cognitivas, se recomienda realizar pruebas neuropsicológicas. Dependiendo de la duración de la enfermedad, hasta el 50% de los pacientes con ELA desarrollan síntomas disejecutivos, y en alrededor del 15% puede diagnosticarse una demencia frontotemporal. Esta observación contradice doctrinas anteriores que no veían alteraciones cognitivas en la ELA. Por cierto, los pacientes con demencia frontotemporal también deben ser sometidos a una búsqueda muy minuciosa de signos de enfermedad de la motoneurona, que también se presenta con mayor frecuencia en el curso de la enfermedad. De lo contrario, los diagnósticos complementarios sirven esencialmente para excluir diagnósticos diferenciales [2].
Tratamiento farmacológico
Además, el riluzol es el único fármaco aprobado para el tratamiento de la ELA; se recomienda prescribirlo lo antes posible en el curso de la enfermedad. El medicamento es seguro; aparte de una leve fatiga, rara vez se produce un aumento de las enzimas hepáticas. El curso de la enfermedad puede ralentizarse algo con riluzol. Además, pueden utilizarse otras terapias farmacológicas orientadas a los síntomas, como la amitriptilina o las gotas de atropina para la salivación alterada. Los fármacos potencialmente sedantes como las benzodiacepinas y los opiáceos pueden utilizarse muy bien para tratar la ansiedad, el dolor y la disnea, sujetos a límites terapéuticos relativamente estrechos.
Nutrición
La pérdida de peso es un marcador pronóstico negativo que debe evitarse si es posible. Los pacientes y sus familiares pueden ser asesorados en consecuencia y se les pueden ofrecer suplementos nutricionales. La estrecha vigilancia logopédica para la detección precoz de la disfagia es de gran importancia para que la oferta de nutrición enteral percutánea mediante una sonda PEG pueda realizarse lo antes posible. Cuanto antes se inserte una PEG en el curso de la enfermedad, menor será el riesgo de complicaciones y más probable será que la medida contribuya a mejorar la calidad de vida y el tiempo de supervivencia. Es importante para los afectados el hecho de que la ingesta adicional de alimentos por vía oral sigue siendo posible de forma habitual.
Ventilación
En cualquier momento del curso de la enfermedad, esté atento a los síntomas de hipoventilación, especialmente por la noche. Además de la disnea, incluyen somnolencia diurna, cefalea matutina u ortopnea. Estos síntomas pueden tratarse muy eficazmente con ventilación domiciliaria no invasiva, que mejora significativamente la calidad de vida de los afectados. Este eficaz tratamiento sintomático debería comentarse con todas las personas afectadas en una fase temprana, cuando aparecen los síntomas correspondientes, ya que puede mejorar la calidad de vida de los afectados, al menos durante un tiempo limitado, como casi ninguna otra terapia [9]. La ventilación invasiva, por otra parte, sólo se considera para un grupo seleccionado de pacientes. Si en algún momento del curso de la enfermedad el paciente desea poner fin a la ventilación, esto debe respetarse y acompañarse en consecuencia. Por regla general, esta medida conduce pronto a laretención de CO2 y los pacientes mueren en unaanestesia de CO2 de lenta instauración.
Terapia adicional
Un tratamiento estrechamente especializado y multiprofesional, como el que se ofrece en un centro de ELA o en instituciones comparables, es esencial para el cuidado óptimo de los pacientes con ELA. Por un lado, las terapias mencionadas pueden aplicarse en una fase temprana y coordinarse de forma óptima y, por otro, pueden complementarse de forma útil otras medidas terapéuticas como la fisioterapia, la terapia ocupacional y la logopedia, cuyo objetivo es preservar las funciones conservadas, como la capacidad de comunicación, durante el mayor tiempo posible.
Literatura:
- Schweikert K: Esclerosis lateral amiotrófica. Foro médico suizo 2015; 15: 1068-1073.
- Andersen PM, et al: Directrices de la EFNS sobre el tratamiento clínico de la esclerosis lateral amiotrófica (ELAM) – informe revisado de un grupo de trabajo de la EFNS. Eur J Neurol 2012; 19: 360-375.
- Brooks BR, et al: El Escorial revisitado: criterios revisados para el diagnóstico de la esclerosis lateral amiotrófica. Amyotroph Lateral Scler Other Motor Neuron Disord 2000; 1: 293-299.
- Carvalho MD, et al: El algoritmo diagnóstico Awaji aumenta la sensibilidad de los criterios de El Escorial para el diagnóstico de la ELA. Esclerosis lateral amiotrófica 2009; 10: 53-57.
- Schulte-Mattler WJ, et al: MUNIX – un biomarcador prometedor en la ELA. Clin Neurophysiol 2015; 46: 186-189.
- Schreiber S, et al: Estado de la experiencia e importancia de los métodos de imagen en los cambios neuromusculares inducidos por la ELA. Clin Neurophysiol 2016; 46: 173-181.
- Felbecker A, et al.: Cuatro pedigríes familiares de ELA discordantes para dos mutaciones de SOD1: ¿son patogénicas todas las mutaciones de SOD1? J Neurol Neurosurg Psychiatry 2010; 81: 572-577.
- Rohrer JD, et al: Expansión de C9orf72 en la demencia frontotemporal y la esclerosis lateral amiotrófica. Lancet Neurol 2015; 14: 291-301.
- Boentert M, et al: Manejo de la ventilación y la secreción en la esclerosis lateral amiotrófica. Clin Neurophysiol 2015; 46: 163-172.
InFo NEUROLOGÍA Y PSIQUIATRÍA 2016; 14(2): 26-29.