La enfermedad pulmonar intersticial (EPI) puede producirse como complicación de varias enfermedades del tejido conectivo, y sus consecuencias son una mayor mortalidad y morbilidad. Se cree que la enfermedad pulmonar intersticial está provocada por una inflamación mediada por el sistema inmunitario y el consiguiente daño arquitectónico de los pulmones. Investigadores británicos han centrado su atención en las opciones de tratamiento para pacientes con esclerosis sistémica, artritis reumatoide y miopatías inflamatorias.
Las opciones farmacoterapéuticas se dirigen principalmente a la inmunosupresión, pero la terapia antifibrótica también se ha mostrado prometedora recientemente. También se utilizan los glucocorticoides, pero sólo deben administrarse en dosis bajas y durante el menor tiempo posible debido a los perfiles de efectos secundarios, escriben el Dr. Zhe Wu y el Dr. Philip Molyneaux, ambos del Instituto Nacional del Corazón y el Pulmón, el Imperial College de Londres y los Hospitales Royal Brompton y Harefield de Londres [1].
Esclerosis sistémica
De todas las enfermedades del tejido conectivo, la EPI se da con mayor frecuencia en la esclerosis sistémica (SSc) (70-90%) y es la causa más común de muerte en ella. El diagnóstico por imagen revela a menudo un patrón inespecífico de neumonía intersticial (NSIP), con una proporción significativa de pacientes que desarrollan hipertensión pulmonar.
En la SSc, la extensión de la fibrosis determina el pronóstico, y sólo los pacientes con enfermedad de moderada a grave requieren tratamiento. Si la tomografía computarizada de alta resolución (TCAR) muestra una extensión de la enfermedad superior al 30% del pulmón o una afectación entre el 10-30% con una capacidad vital forzada (CVF) <70%, el médico debe tomar medidas terapéuticas. En el pasado, aquí se utilizaban sobre todo los glucocorticoides. Sin embargo, el uso prolongado de esteroides en dosis superiores a 10 mg de prednisolona al día se asocia a un mayor riesgo de crisis renal y debe evitarse.
Varios ensayos controlados aleatorizados han abordado el enfoque del tratamiento con agentes ahorradores de esteroides: el estudio multicéntrico Scleroderma Lung Study I (SLS I) demostró que la ciclofosfamida oral (CYC) era eficaz en 1-2 mg por kgKG/día tuvo un impacto modesto pero estadísticamente significativo en la CVF y la capacidad pulmonar total (CPT) tras un año de tratamiento en comparación con el placebo (mejora media del 2,5% y el 4,1%, respectivamente). También se observaron mejoras clínicamente relevantes con respecto a la disnea, los cambios cutáneos y el estado funcional. Además, las tomografías computarizadas de los pacientes del brazo placebo tenían más probabilidades de mostrar fibrosis progresiva. Sin embargo, el beneficio de la CVF no persistió un año después de finalizar el tratamiento.
El posterior ensayo SLS-II comparó el tratamiento con CYC oral durante 12 meses con 24 meses de micofenolato mofetil (MMF). La dosis diaria objetivo de MMF fue de 3 g. Con la misma eficacia, el MMF fue mucho mejor tolerado, con un riesgo significativamente menor de desarrollar leucopenia y trombocitopenia.
EULAR recomienda el CYC
El ensayo FAST utilizó dosis bajas de esteroides en combinación con seis infusiones intravenosas de CYC (con una dosis de 600 mg/m2 al mes), seguido de una terapia de mantenimiento con azatioprina (2,5 mg/kgKG al día), y mostraron efectos positivos sobre la CVF y la extensión radiológica de la enfermedad, aunque estos resultados no alcanzaron significación estadística, posiblemente debido al pequeño tamaño de la muestra, escriben los autores. Además, la cohorte estudiada mostró una menor disfunción pulmonar en comparación con el grupo SLS-I. Sorprendentemente, según el Dr. Wu y el Dr. Molyneaux, el perfil de efectos secundarios del régimen intravenoso de CYC parecía ser más favorable que el de la administración oral. El impacto de esto es importante en el entorno del mundo real, donde el cumplimiento variable o incluso la interrupción del tratamiento son habituales. Como resultado de estos estudios, la Liga Europea contra el Reumatismo (EULAR) recomienda el uso de CYC en pacientes con empeoramiento de la enfermedad pulmonar.
En cuanto al uso de antifibróticos en la fibrosis pulmonar, el ensayo SENSCIS investigó la eficacia y seguridad del nintedanib en la SSc-ILD en comparación con la terapia estándar. En el estudio participaron 576 sujetos con más de un 10% de fibrosis, de los que aproximadamente la mitad recibieron micofenolato mofetilo al inicio del estudio. La reducción relativa del descenso de la CVF con nintedanib fue del 44%, similar a los índices observados previamente en ensayos de FPI. Sin embargo, no se observó ningún efecto del tratamiento sobre la disnea, la calidad de vida o las manifestaciones cutáneas. El nintedanib fue bien tolerado, y más de cuatro quintas partes de los pacientes del brazo activo completaron toda la duración del tratamiento. Posteriormente, un análisis post hoc demostró que el nintedanib tenía efectos similares sobre la reducción relativa del descenso de la CVF tanto en el grupo de MMF como en el de no MMF (40% y 46%, respectivamente). Sin embargo, el momento óptimo para la introducción de la terapia antifibrótica sigue siendo cuestionable. Ensayos más pequeños de fase temprana de pirfenidona en SSc-ILD han mostrado ahora una seguridad y tolerabilidad favorables, lo que ha conducido al ensayo SLS-III actualmente en curso, que evaluará el efecto de una combinación de MMF y pirfenidona en comparación con la monoterapia con MMF.
Entre los biológicos, el rituximab (RTX) se ha mostrado prometedor en varios ensayos pequeños. El ECA multicéntrico RECITAL comparó el RTX (dos dosis con 14,5 días de intervalo) con infusiones mensuales intravenosas de CYC (seis dosis) para el tratamiento de una serie de CTD, incluida la SSc. Se esperan los resultados al respecto. El agente anti-interleucina (IL)-6 tocilizumab ha demostrado un efecto estabilizador potencial sobre la CVF en dos ECA (faSScinate y focuSSced), aunque en la inscripción no se examinó la presencia de EPI al inicio del estudio. Por lo tanto, los participantes presentaban un deterioro mínimo de la función pulmonar, y la eficacia de los fármacos anti-IL-6 en pacientes con EPI de moderada a grave requiere una evaluación adicional, señalaron los autores.
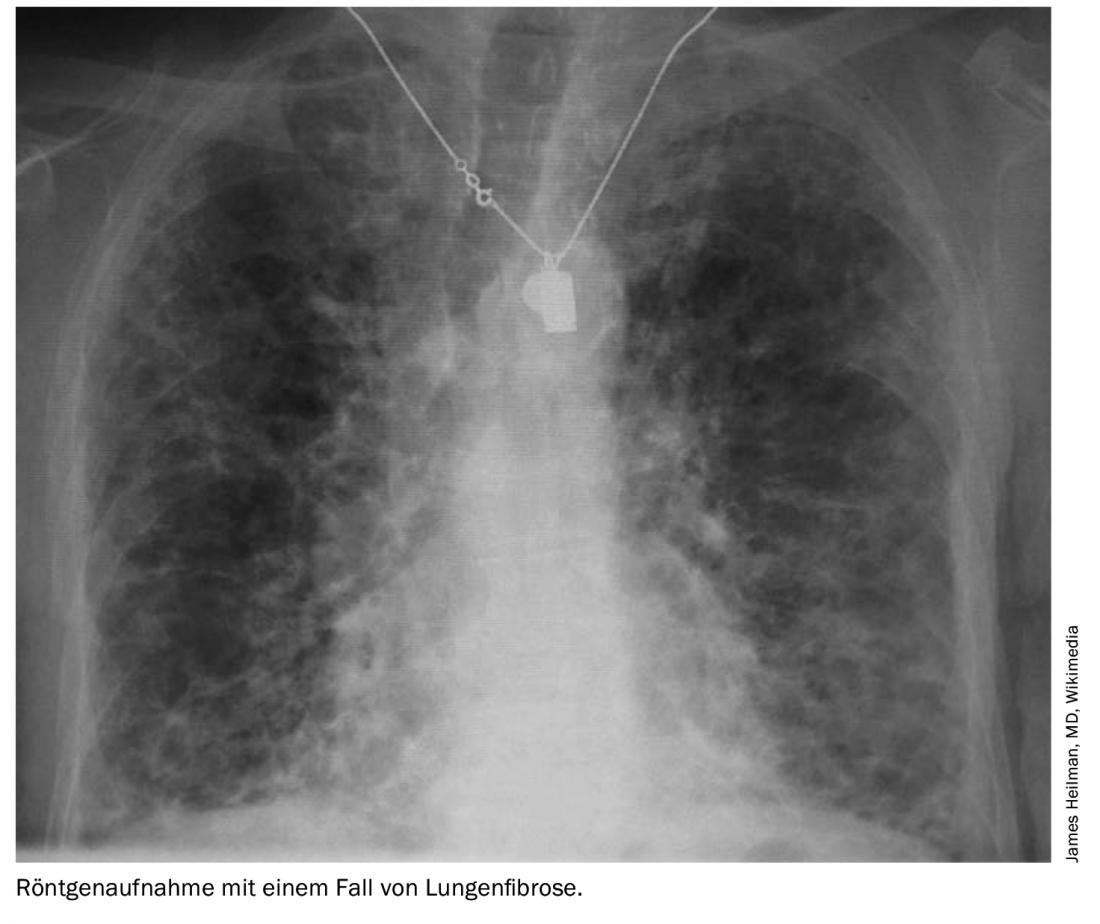
Artritis reumatoide
Se calcula que la prevalencia de la enfermedad pulmonar intersticial en la artritis reumatoide (AR) es del 5-10%, y que la cifra aumenta a medida que aumenta el cribado de pacientes con AR. El patrón de TC predominante es la neumonía intersticial habitual (NIU), seguida de la NSIP, siendo la primera la que ofrece un peor pronóstico. Existe un consenso general en que los esteroides deben ser los fármacos de primera elección. La literatura actual no aporta pruebas concluyentes sobre la terapia ahorradora de esteroides, aunque el RTX ha mostrado algunos resultados prometedores, escriben Wu y Molyneaux.
El estudio INBUILD investigó la eficacia del nintedanib en varios subtipos de EPI con fenotipo progresivo. Aunque el estudio no se diseñó para analizar específicamente el efecto en pacientes con AR, la señal general fue positiva. Por lo tanto, parece probable que los pacientes con AR y un cuadro UIP más fibrótico reciban en el futuro un tratamiento antifibrótico en una fase más temprana de la evolución de su enfermedad.
Una controversia ya histórica en el tratamiento de la CTD-ILD es la relación entre el metotrexato (MTX) y la EPI en pacientes con AR: Algunas publicaciones antiguas de baja calidad sugieren que el MTX está asociado a la EPI fibrótica, pero estos estudios se realizaron antes del uso de la tomografía computarizada. Estudios mucho más amplios y sólidos han refutado recientemente esta asociación y sugieren que el uso de MTX puede retrasar la aparición de la enfermedad pulmonar intersticial. Un estudio contrastó la exposición al MTX en pacientes con AR con EPI (n=410) frente a pacientes sin EPI (n=673) y demostró que el uso de MTX se asociaba a un menor riesgo de desarrollar AR-ILD (OR 0,43; IC del 95%: 0,26-0,69). Este hallazgo se confirmó en un estudio de cohortes prospectivo multicéntrico de más de 2000 pacientes con AR recién diagnosticada. Otros estudios han corroborado estos hallazgos, por lo que el MTX ya no debería suspenderse de forma rutinaria en pacientes con artritis reumatoide con enfermedad articular bien controlada que desarrollen EPI, concluyen los autores.
Miopatías inflamatorias idiopáticas
Las miopatías inflamatorias idiopáticas (MII) son un grupo de trastornos que incluyen la polimiositis, la dermatomiositis y el síndrome antisintetasa (SAS). Los patrones de EPI más comunes son la NSIP y la neumonía organizativa, que a menudo pueden darse simultáneamente. Los pacientes con el subtipo de anticuerpos anti-MDA5 suelen desarrollar un fenotipo clínicamente amiopático y rápidamente progresivo parecido a la neumonía intersticial aguda. A menudo requieren un enfoque terapéutico agresivo y combinado similar al tratamiento de la neumonía intersticial aguda.
Como en otros CTD-ILD, los esteroides son la terapia de primera línea, pero las recaídas son frecuentes y a menudo es necesario añadir una terapia de segunda línea para lograr la remisión. Una revisión sistemática concluyó que la administración intravenosa de CYC durante 6-12 meses mejoraba la función pulmonar y el aspecto del TAC en más de la mitad de los pacientes y restablecía la fuerza muscular en cuatro quintas partes. El rituximab se ha mostrado prometedor en un pequeño estudio piloto de 10 sujetos con TEA que recayeron tras la terapia inmunosupresora inicial: En la mayoría de los pacientes (9 de cada 10), el rituximab estabilizó o mejoró la función pulmonar. Otra serie de casos retrospectiva monocéntrica que examinó la eficacia a largo plazo del RTX en 34 pacientes con ASA (seguimiento medio de 52 meses) informó de una mejora del 24% en la mediana de la CVF y del 17% en la capacidad de difusión del pulmón para el monóxido de carbono.
El inhibidor de la calcineurina tacrolimus ha demostrado su eficacia como terapia añadida a los esteroides y también en combinación con otros inmunosupresores. Sin embargo, las pruebas no son claras y, dada la elevada mortalidad, existe una necesidad urgente de descubrir tratamientos para esta cohorte, afirman los autores. Concluye que, en general, es esencial un seguimiento estrecho de la función pulmonar para guiar el inicio del tratamiento y evaluar la respuesta. El uso de antifibróticos es un campo prometedor, pero plantea más preguntas sobre el momento de inicio del tratamiento y si se utiliza con inmunosupresión concurrente o como terapia independiente.
Literatura:
- Wu Z, Molyneaux PL: Elección de la farmacoterapia para la EPI en pacientes con enfermedad del tejido conectivo. Breathe 2021; 17: 210114; doi: 10.1183/20734735.0114-2021.
InFo DOLOR Y GERIATURA 2022; 4(1-2): 26-27