Los defectos congénitos del corazón son la malformación orgánica más frecuente en los niños. Todo defecto cardíaco requiere una evaluación y clasificación cardiológica pediátrica. El seguimiento cardiológico pediátrico especializado es esencial teniendo en cuenta el crecimiento del niño, los posibles hallazgos cardiacos residuales tras la intervención/cirugía y cualquier complicación tardía a lo largo de la infancia y la adolescencia.
Los defectos congénitos del corazón son la malformación orgánica más común, y se dan en alrededor del 1% de todos los recién nacidos. Para Suiza, esto correspondió a aproximadamente 850 niños con 85 287 nacidos vivos en 2014. El espectro va desde los defectos cardiacos simples que apenas afectan al sistema cardiovascular hasta los graves (Tab. 1) que pueden provocar una muerte prematura si no se tratan [1].

La frecuencia con la que se producen los distintos defectos cardiacos varía enormemente (Tab. 2) y cada defecto cardiaco requiere una evaluación y clasificación cardiológica pediátrica. El método de examen diagnóstico más importante es – aparte del examen clínico que incluye la exploración del paciente – el examen del paciente. Auscultación – ecocardiografía, que permite visualizar y definir prácticamente todas las estructuras cardiacas.

El electrocardiograma (ECG) (también ECG Holter 24h) detecta cualquier arritmia o anomalía de conducción, así como signos de hipertrofia. Dependiendo de la situación, están indicados otros métodos de examen especiales como la espiroergometría, la resonancia magnética cardiaca (RMc), la tomografía computerizada (TC) o el cateterismo cardiaco para diagnósticos invasivos (por ejemplo, medición de la presión, angiografías). Este último también ofrece la posibilidad de intervenir al mismo tiempo. Esto significa que hoy en día la mayoría de los defectos cardíacos se diagnostican en el primer año de vida. Sin una terapia quirúrgica o intervencionista, la esperanza de vida se reduce considerablemente en el caso de los defectos cardiacos moderados y graves.
Los extraordinarios avances logrados en cirugía cardiaca, cardiología intervencionista, anestesia y medicina intensiva en las últimas décadas han mejorado significativamente las posibilidades de supervivencia tras una intervención cardiaca en la infancia. Por ejemplo, más del 90% de los niños con defectos cardíacos congénitos llegan ahora a la edad adulta [2,3]. Sin embargo, a menudo se enfrentan a problemas polifacéticos a lo largo de su vida debido a su fisiopatología cardiaca y a las enfermedades concomitantes que a menudo también presentan. Un punto clave es que la mayoría de los defectos cardiacos pueden repararse, pero no corregirse en sentido estricto [4].
Los hallazgos cardiacos residuales significativos y el crecimiento del niño hacen imprescindible un seguimiento cardiológico pediátrico especializado. Un seguimiento óptimo de los niños con cardiopatías requiere una estrecha colaboración entre los médicos generalistas y pediatras de la práctica privada y los médicos especialistas implicados. Esto es especialmente importante en el caso de enfermedades concomitantes complejas que perjudican el desarrollo neurológico, por ejemplo [5].
En la adolescencia, la transición a la medicina de adultos a un cardiólogo con formación específica (cardiólogo GUCH; GUCH = Grown-Ups with Congenital Heart Disease) debe iniciarse a tiempo en el sentido de un flujo continuo de información [6].
Complicaciones en el curso
Arritmias: Algunos defectos cardíacos están asociados a arritmias precoces. Por ejemplo, puede producirse un bloqueo auriculoventricular completo con la transposición cc de las grandes arterias. Sin embargo, en la mayoría de los casos, las arritmias se producen debido a residuos hemodinámicos, como una carga de presión persistente con fibrosis ventricular o carga de volumen y/o cicatrización miocárdica (atrio-ventriculotomía), tras la cirugía cardiaca. Un trastorno del ritmo, incl. Trastorno de la conducción, puede desarrollarse al principio o al final del postoperatorio. Las observaciones incluyen taquicardia supraventricular (normalmente taquicardia por reentrada), aleteo o fibrilación auricular, taquicardia auricular ectópica, síndrome del seno enfermo, taquicardia ventricular, bloqueo auriculoventricular y muerte súbita cardiaca.
Mientras que el riesgo de arritmia sigue siendo bajo tras operaciones cardiacas más sencillas, aumenta significativamente tras operaciones complejas como la paliación de Fontan en corazones univentriculares. Por esta razón, el ECG (a largo plazo) y la ergometría forman parte de la evaluación médica regular de estos pacientes. Para las arritmias que se producen en el postoperatorio tardío, hoy en día se dispone de la electrofisiología invasiva, además de la terapia farmacológica, que consigue buenos resultados mediante la ablación de, por ejemplo, las cicatrices intracardiacas.
Limitaciones físicas: Con el creciente número de niños que llegan a la edad adulta tras una reparación o paliación quirúrgica, la inclusión de los niños en el tejido social con sus iguales se ha convertido en un objetivo importante. Mientras que un paciente cardiosaludable puede quintuplicar su gasto cardiaco durante el ejercicio, un paciente con un defecto cardiaco complejo, como tras una paliación Fontan en el corazón univentricular, como mucho puede duplicarlo. El resultado es una resistencia física limitada [7].
En general, se apoya la actividad deportiva en el ámbito de la resistencia. Por el contrario, el entrenamiento isométrico con pesas o los deportes de competición no son recomendables para la mayoría de los pacientes.
Las pruebas y la evaluación del rendimiento físico mediante espiroergometría son un componente habitual de los exámenes de seguimiento cardiológico pediátrico a partir de los diez años de edad aproximadamente.
Retraso en el desarrollo/desarrollo neurocognitivo: Los niños con un defecto cardiaco grave pueden desarrollar un retraso en el desarrollo y entonces son más pequeños y de estatura más larguirucha que los niños sanos de la misma edad. Diversos estudios han demostrado que los niños con defectos cardiacos complejos tienen un mayor riesgo de sufrir déficits neurocognitivos tras una intervención quirúrgica cardiaca en la etapa neonatal o infantil. El retraso en el desarrollo de la motricidad fina y gruesa, los problemas de comportamiento, la atención reducida, los síntomas de hiperactividad y el retraso en el lenguaje son déficits comunes [6,8]. Esto puede tener un impacto negativo en la vida cotidiana y en las carreras educativas y profesionales en la edad adulta. Los niños con defectos cardíacos de leves a moderados no suelen verse afectados. Es importante plantear estas cuestiones a los padres y ofrecerles apoyo específico con el pediatra o el médico de cabecera.
Endocarditis: La endocarditis infecciosa es una complicación grave y potencialmente mortal en pacientes con cardiopatías congénitas o cardiopatías adquiridas. Por ello, la educación de padres y pacientes es de gran importancia durante las revisiones cardiológicas pediátricas. Los síntomas de endocarditis deben reconocerse pronto para poder dar una respuesta adecuada. También deben comunicarse los beneficios y la correcta administración de la profilaxis de la endocarditis con medicamentos. Las directrices actuales para la profilaxis de la endocarditis en niños en Suiza se enumeran en la tabla 3.

Hipertensión arterial pulmonar: Todos los defectos cardíacos congénitos con grandes derivaciones intra o extracardíacas conducen a una carga de volumen y presión sin restricciones del lecho vascular pulmonar. Como resultado, puede desarrollarse una hipertensión arterial pulmonar que provoque cambios irreversibles en las últimas fases de esta grave enfermedad. Por ejemplo, en el caso de una comunicación interventricular grande no corregida con derivación izquierda-derecha, se produce un aumento de la presión en el lecho vascular pulmonar a lo largo de los años, que acaba en una inversión de la derivación con cianosis debido a una nueva derivación derecha-izquierda (reacción de Eisenmenger). Uno de los objetivos de la intervención quirúrgica en la primera infancia es prevenir este desarrollo. Los pacientes con defectos cardíacos congénitos tienen un mayor riesgo de desarrollar hipertensión arterial pulmonar incluso sin la presencia de shunts relevantes. Dependiendo de su gravedad, esto tiene un efecto perjudicial sobre la calidad y la duración de la vida [10]. Para el diagnóstico y el inicio de una terapia específica, suele ser necesario estar conectado a un centro especializado.
Enfermedades concomitantes: Los defectos cardíacos congénitos están asociados a otras malformaciones y/o anomalías cromosómicas en un 15-20%. Los niños con trisomía 21 presentan vitium cordis en el 40-50% de los casos. Estos pacientes, a menudo con malformaciones complejas, se benefician de una atención multidisciplinar.
Ejemplos de cuidados de seguimiento y aspectos específicos de defectos cardíacos individuales
Estenosis del istmo aórtico (AIST): El objetivo es eliminar la estenosis en una fase temprana y crear una aorta lo más libre de gradientes y de calibre normal posible. Esto sólo puede conseguirse mediante cirugía o cateterismo cardíaco intervencionista (dilatación con balón o implantación de un stent) (Fig. 1). El método de tratamiento depende de la edad del paciente, así como de la morfología del AIST. A menudo hay malformaciones asociadas (por ejemplo, válvula aórtica bicúspide en aprox. 50% de los casos).
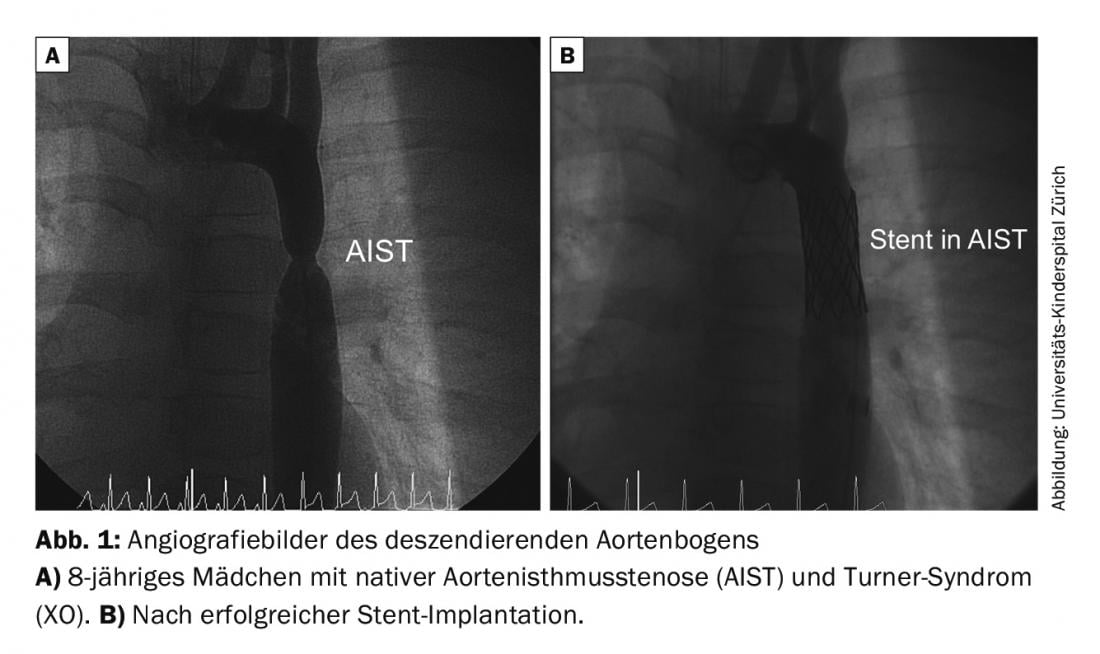
Los pacientes con reparación quirúrgica tienen un riesgo del 5-10% de reestenosis en el curso. El riesgo de hipertensión arterial aumenta con la edad, incluso si la estenosis se ha eliminado eficazmente. Otros posibles riesgos son el desarrollo de aneurismas/disecciones aórticas en el lugar de la sutura o en la endoprótesis.
Recomendaciones de seguimiento [11]:
- Por lo general, anualmente, en el transcurso de al menos un año. cada 2 años:
- estado clínico
- Medición de la presión arterial (PA) en las 4 extremidades
- en función de la PA en reposo: medición de la PA en 24h
- ECG
- Ecocardiografía (tamaño del ventrículo izquierdo, grosor y función del miocardio, grado de estenosis sobre el arco aórtico/estroma, preste más atención a las malformaciones asociadas si es necesario)
- Ergometría (a partir de 10 años) cada 3-4 años
- RMNc: en caso de visualización insuficiente en la ecocardiografía: visualización del arco aórtico, exclusión de la formación de aneurismas cada 5 años
d-transposición de las grandes arterias (d-TGA): Hoy en día, la cirugía de switch arterial para restablecer la concordancia ventrículo-arterial de la d-TGA es el tratamiento de primera elección. La aorta y la arteria pulmonar se intercambian y mediante la llamada maniobra de Lecompte, la bifurcación pulmonar se sitúa finalmente delante de la aorta (Fig. 2) . Al mismo tiempo, es necesario reimplantar las arterias coronarias. La válvula pulmonar anatómica sirve así de por vida como válvula neoaórtica en la circulación sistémica.
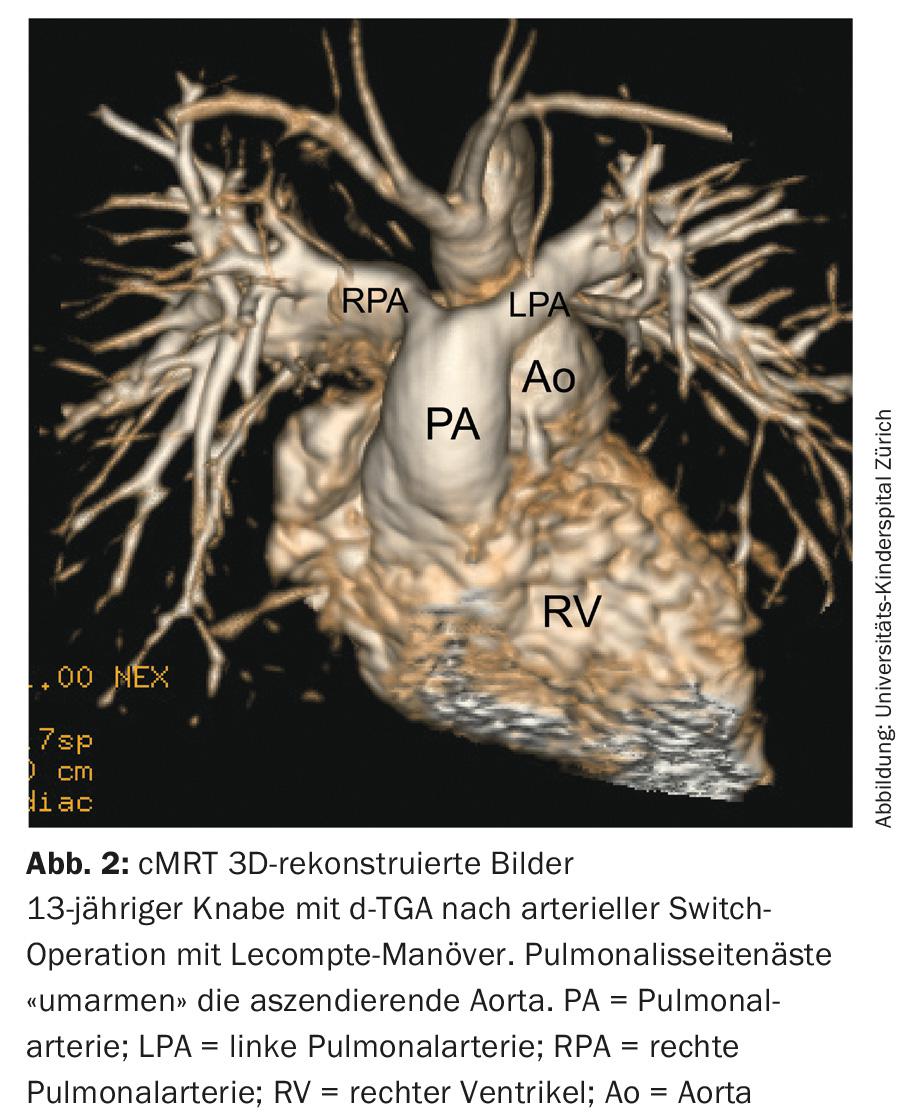
A largo plazo, esta válvula neoaórtica puede volverse disfuncional y la raíz aórtica dilatarse. Debido a la maniobra de Lecompte con desplazamiento de las ramas colaterales pulmonares anteriores a la aorta ascendente, puede producirse torsión y estiramiento y, por tanto, estenosis de las ramas colaterales pulmonares. Como la operación implica la transferencia coronaria, la torsión y el estiramiento también pueden provocar la estenosis o, en casos extremos, la oclusión de una arteria coronaria, con el consiguiente infarto de miocardio y muerte súbita cardiaca.
Recomendaciones de seguimiento [9,11]:
- Normalmente anualmente, en el transcurso de al menos un año. cada 2 años:
- estado clínico
- ECG
- Ecocardiografía (tamaño y función ventricular, grado de estenosis de las ramas laterales pulmonares, trastorno de la movilidad de la pared, función de la válvula neo aórtica, dimensión de la raíz neo aórtica).
- ECG a largo plazo según evaluación individual
- Angiografía coronaria en curso perioperatorio complicado, paciente sintomático o diagnóstico rutinario anormal.
- Ergometría (a partir de 10 años) cada 2-3 años
- RMNc: en caso de mala calidad del sonido, para la evaluación del tamaño y la función ventricular derecha e izquierda y la evaluación de las ramas laterales pulmonares. Evaluación de la arteria coronaria y/o cicatrización miocárdica.
Tetralogía de Fallot (TOF): El objetivo de la reparación quirúrgica anatómica de la tetralogía de Fallot es el cierre del parche de la CIV y la eliminación de la obstrucción del tracto de salida del ventrículo derecho (OVVD). Si el anillo de la válvula pulmonar tiene un diámetro pequeño y/o la válvula es displásica, es necesario implantar un parche transanular, lo que siempre conduce a una insuficiencia de la válvula pulmonar (Fig. 3) . A medio y largo plazo, esto da lugar a una dilatación relevante del ventrículo derecho, que puede provocar disfunción ventricular, deterioro del rendimiento y arritmia y, en última instancia, hace necesaria la sustitución de la válvula pulmonar.

El bloqueo completo de rama derecha es frecuente tras el cierre con parche de la CIV; el riesgo de bloqueo auriculoventricular completo que requiera la implantación de un marcapasos es pequeño (<2%).
Recomendaciones de seguimiento [9,11]:
- Normalmente anualmente
- estado clínico
- ECG (anchura del QRS)
- Ecocardiografía (RVOTO residual incl. ramas laterales pulmonares, regurgitación de la válvula pulmonar, tamaño y función del ventrículo derecho, CIV residual, tamaño y función del ventrículo izquierdo, dimensión de la raíz aórtica).
- ECG de larga duración (taquicardia no sostenida) min. cada 3 años en pacientes asintomáticos
- Ergometría (a partir de 10 años), cada 3-5 años
- RMNc: con dilatación creciente del VD: para evaluar el tamaño y la función del ventrículo derecho y valorar la insuficiencia pulmonar (fracción de regurgitación), el tamaño y la función del ventrículo izquierdo. Evaluación de las ramas laterales pulmonares.
Ventrículo único (SV): En el caso de los corazones univentriculares, se distinguen dos formas básicas, dependiendo de si el ventrículo derecho o el izquierdo no se han formado lo suficiente (por ejemplo, atresia tricúspide o síndrome del corazón izquierdo hipoplásico); Fig. 4). La terapia quirúrgica en forma de tres intervenciones paliativas generalmente escalonadas termina en la “circulación de Fontan”. El objetivo es crear un circuito conectado en serie a partir de los circuitos paralelos existentes hasta ahora. En el periodo neonatal, el primer paso consiste en estabilizar la circulación paralela, lo que, dependiendo del defecto cardíaco, suele implicar una derivación o una reducción de la hiperperfusión pulmonar mediante un vendaje. En un segundo paso, se crea una anastomosis cavopulmonar superior bidireccional (Glenn) en la infancia, lo que provoca una descompresión significativa del volumen del corazón. Con la finalización del Fontan a los 2-3 años de edad mediante conducto extracardiaco, anastomosis cavopulmonar total, se consigue la paliación definitiva en pacientes con corazón univentricular. Sólo con esta separación final de la circulación se levanta la cianosis (aparte de las pequeñas derivaciones residuales; colaterales o fenestración).
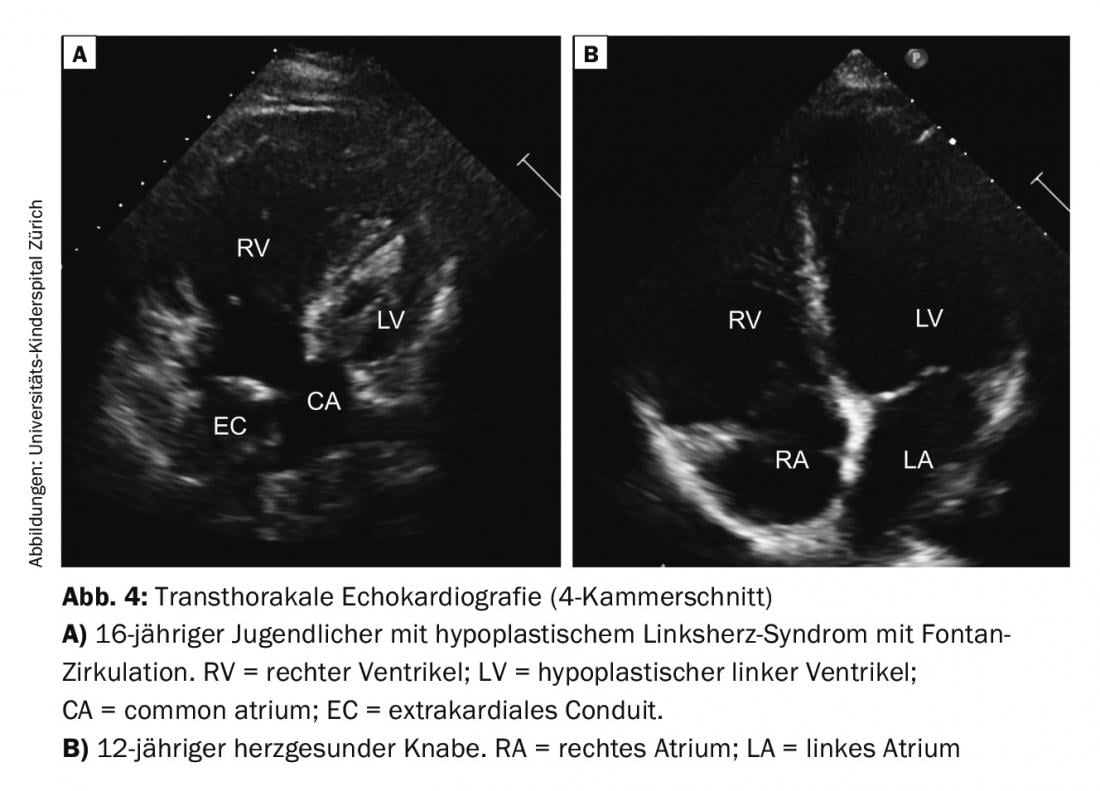
La atención durante los pasos quirúrgicos individuales y el seguimiento de este grupo heterogéneo y único de pacientes con todas sus complicaciones y efectos tardíos es un gran reto para los cardiólogos pediátricos y posteriormente para los cardiólogos especializados en adultos (cardiólogo GUCH).
Las complicaciones en estos pacientes son muy diversas: función ventricular reducida, insuficiencia de la válvula AV, estenosis en la circulación sistémica o pulmonar, arritmia, incompetencia cronotrópica, trombosis/embolia, enteropatía perdedora de proteínas, bronquitis plástica, disfunción hepática, hipertensión pulmonar, paresia diafragmática, escoliosis, etc.
Recomendaciones para el seguimiento tras la finalización de la fontanela [9,11]:
- min. anual
- estado clínico
- Medición de la PA en las 4 extremidades
- Saturación transcutánea de oxígeno
- ECG
- Ecocardiografía (función del SV, insuficiencia de la válvula AV, obstrucción izquierda (estenosis subaórtica, estenosis ístmica), perfil de flujo de la anastomosis cavopulmonar superior o en la circulación de Fontan y las venas hepáticas, etc.).
- Examen de laboratorio: proteínas totales o albúmina sérica, valores hepáticos, pro-BNP como parámetro de la función cardiaca, hemoglobina.
- ECG de larga duración (taquicardia reentrante intraauricular no sostenida, competencia cronotrópica), si hay indicios de arritmia o al menos algunos signos de infarto. todo
- 3 años
- Ergometría (a partir de 10 años), min. Cada 3 años
- IRMc: Evaluación de la función ventricular, cuantificación del flujo sanguíneo, visualización anatómica de las conexiones de Fontan. Su aplicación depende del curso clínico, pero suele recomendarse en la adolescencia y antes de la transición a los cardiólogos de la GUCH.
- Cateterismo cardíaco de diagnóstico: en caso de deterioro de la circulación de Fontan, como disminución de la capacidad de ejercicio, aumento de la cianosis, ascitis o enteropatía perdedora de proteínas, bronquitis plástica, etc.
Palabras finales
Hoy en día, la mayoría de los defectos cardiacos congénitos tienen un buen pronóstico a largo plazo. La detección precoz del defecto cardiaco, el tratamiento quirúrgico y/o intervencionista por parte de un equipo experimentado y un cuidadoso seguimiento contribuyen decisivamente a ello. Es esencial una estrecha colaboración entre los pediatras y los cardiólogos pediátricos, así como con otros especialistas si es necesario, y la posterior transición a un cardiólogo GUCH.
Literatura:
- Hoffmann JIE, et al: La incidencia de las cardiopatías congénitas. JACC 2002; 39(12): 1890-1900.
- Warnes CA, et al: Grupo de trabajo 1: El perfil cambiante de las cardiopatías congénitas en la vida adulta. JACC 2001; 37(5): 1161-1198.
- Moons P, et al: Tendencias temporales en la supervivencia hasta la edad adulta entre los pacientes nacidos con cardiopatías congénitas de 1970 a 1991 en Bélgica. Circulation 2010; 122: 2264-2272.
- Oechslin E: Defectos cardiacos congénitos: atención especializada de por vida para una enfermedad crónica. Medicina cardiovascular 2006; 9: 373-375.
- Mackie A: Niños y adultos con cardiopatías congénitas perdidos durante el seguimiento. Circulation 2009; 120(4): 302-309.
- Bauersfeld U: Transición, traslado y cooperación en pacientes con defectos cardíacos congénitos – colaboración continua de la cardiología pediátrica y de adultos. Medicina cardiovascular 2006; 9: 336-341.
- Gewillig M, et al: Fracaso de la circulación Fontan. Heart Failure Clin 2014; 10(1): 105-116.
- Sterken C, et al: Desarrollo neurocognitivo tras cirugía cardiaca pediátrica. Pediatría 2016; 137(6), doi: 10.1542/peds.2015-4675.
- Wernovsky G, et al: Directrices para el tratamiento ambulatorio de las cardiopatías congénitas complejas. Congenit Heart Dis 2006; 1: 10-26.
- Dimopoulos K, et al: Hipertensión pulmonar relacionada con cardiopatías congénitas: una llamada a la acción. Eur Heart J 2014; 35(11): 691-700.
- Sociedad Alemana de Cardiología Pediátrica e.V., ed. Schmaltz AA: Directrices para el diagnóstico y la terapia en cardiología pediátrica. Elsevier GmbH, Urban & Fischer Verlag 2007.
- Knirsch W, et al: Nuevas recomendaciones para la profilaxis antibiótica de la endocarditis en niños en Suiza. Pediatrica 2009; 20(4): 28-34.
CARDIOVASC 2016; 15(5): 4-9