Si una o ambas cavidades del corazón están excesivamente engrosadas o dilatadas, el corazón ya no es tan eficaz. Las miocardiopatías se caracterizan por una variedad de causas diferentes de dichos cambios en el tejido muscular del corazón. Sin embargo, la diferenciación de las manifestaciones clínicas individuales tiene importantes implicaciones para el pronóstico y los posibles regímenes de tratamiento.
Las miocardiopatías tienen muchas caras. Básicamente, pueden clasificarse en cuatro fenotipos morfológicos: miocardiopatía hipertrófica (MCH), miocardiopatía dilatada (MCD), miocardiopatía arritmogénica (MRA) y miocardiopatía restrictiva (MCR). Sin embargo, esta clasificación aproximada, basada en puntos de vista, tiene bastante poco en común con las causas reales, como se desprende del gran número de subcategorías. El Prof. Dr. med. Benjamin Meder, de Heidelberg (D), demostró que la imagen por sí sola no es suficiente para detectar la enfermedad. En consecuencia, deben añadirse las características funcionales. Ello se debe a que la miocardiopatía es, en última instancia, la descripción de un fenotipo morfológico y funcional del miocardio que no puede explicarse por una enfermedad arterial coronaria o una alteración de las propiedades de llenado debida a la hipertensión arterial, la vitiación valvular o una cardiopatía congénita.
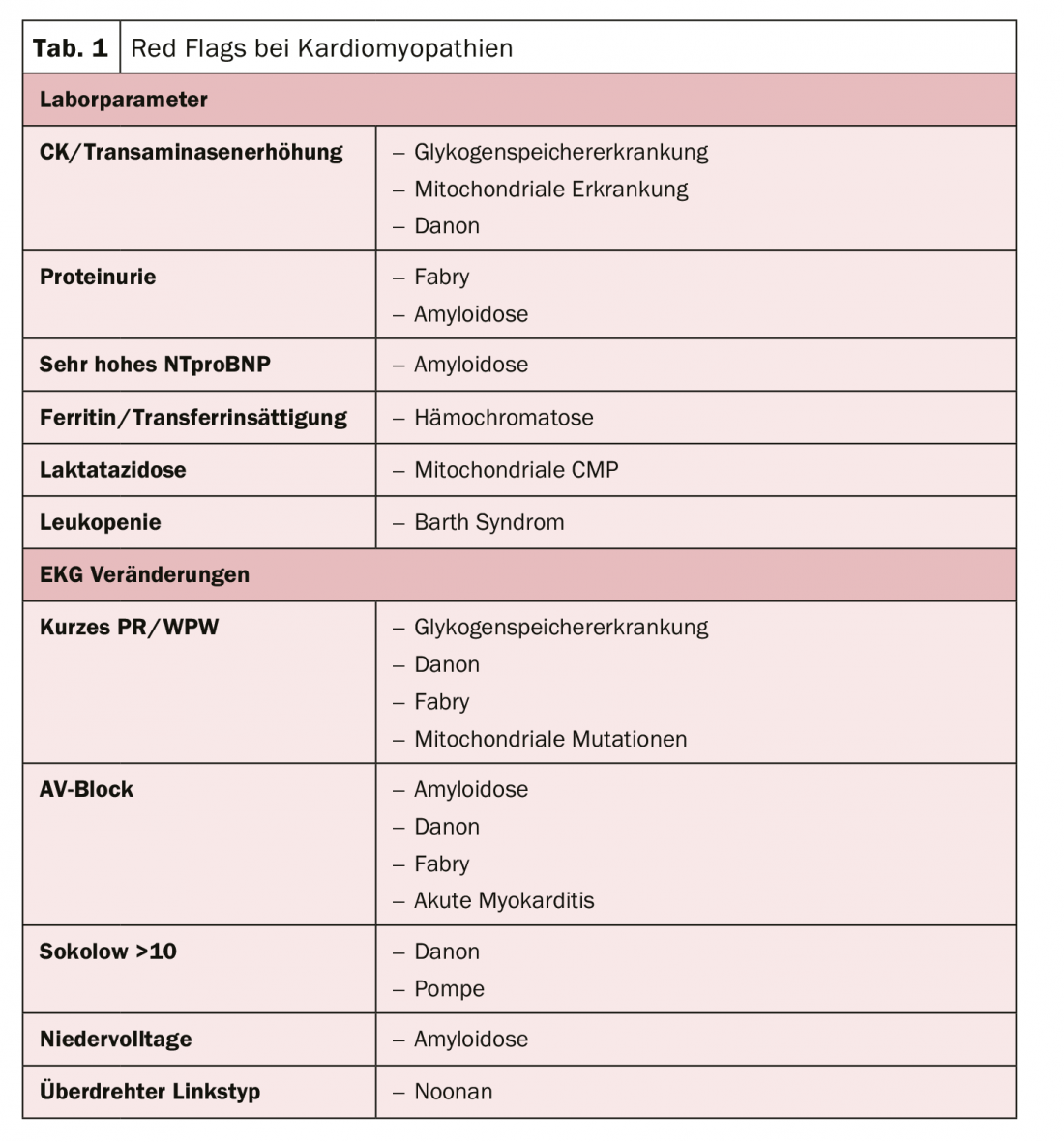
Por tanto, el enfoque estructural del tratamiento optimizado se aproxima a través de la etiología con el objetivo de evitar la muerte súbita cardiaca, el problema limitante de todas las miocardiopatías. Existen parámetros de laboratorio típicos y anomalías en el ECG que pueden indicar la presencia de una miocardiopatía (tab. 1). Ser capaz de hacer estas distinciones es por tanto relevante, ya que el pronóstico viene determinado esencialmente por la causa, afirma el experto. Especialmente los pacientes con amiloidosis tienen una supervivencia muy pobre.
Detectar la amiloidosis y tratarla eficazmente
La amiloidosis puede dividirse en tres tipos en función de la afectación de los órganos: Amiloidosis AL que afecta a los riñones, el corazón y/o los intestinos, amiloidosis mt-ATTR que afecta principalmente al corazón o al sistema nervioso, y amiloidosis wt-ATTR que afecta principalmente al corazón. En principio, la amiloidosis AL puede entenderse como una complicación de la discrasia de células plasmáticas. Por lo tanto, se utiliza una terapia hematológica similar a la del mieloma múltiple con el objetivo de eliminar lo antes posible las cadenas ligeras amiloidógenas y (cardio)tóxicas. Además de la quimioterapia a dosis altas para los pacientes aptos, se utiliza principalmente un régimen basado en inhibidores del proteasoma. En los pacientes más jóvenes, debe favorecerse el CyBorD (bortezomib, ciclofosfamida, dexametasona) frente al BMDex (bortezomib, melfalán, dexametasona) debido a la toxicidad para las células madre del melfalán, con el fin de preservar la opción de una posterior aféresis de células madre y una quimioterapia de dosis alta.
En la amiloidosis causada por depósitos de proteína transtiretina, fármacos como el diflunisal y el tafamidis pueden estabilizar la proteína mutada. Además, las terapias génicas que reducen la producción de transtiretina (por ejemplo, patisiran, inotersen) pueden reducir los efectos sobre el sistema nervioso.
Para los pacientes con AL cardiaca sintomática y amiloidosis ATTR, se aplican en principio las mismas recomendaciones terapéuticas generales que para los pacientes con insuficiencia cardiaca. Sin embargo, incluso dosis bajas de β-bloqueantes o inhibidores de la ECA pueden provocar hipotensión sintomática. Por lo tanto, el tratamiento en pacientes con amiloidosis cardiaca se basa principalmente en la dosis correcta de diuréticos.
Afectación sindrómica de muchos sistemas orgánicos – Enfermedad de Fabry
La enfermedad de Fabry es un trastorno hereditario genéticamente que suele manifestarse entre los 20 y los 40 años. Se forma una enzima α-Gal A mal plegada o no funcional, por lo que se altera el transporte desde el retículo endoplásmico al lisosoma. Esto conduce a una acumulación de sustratos lisosomales. Además del corazón, pueden verse afectados los riñones, el sistema nervioso central, pero también el sistema nervioso periférico y la piel. Si no se trata, la insuficiencia renal es la causa más frecuente de muerte en estos pacientes, explicó la Prof. Dra. med. Ingrid Kindermann, Homburg/Saar (D). Los síntomas cardíacos se dan en más de la mitad de los pacientes de Fabry. Las arritmias malignas son las principales responsables de la muerte súbita cardiaca.
Durante mucho tiempo, la terapia se limitó a aliviar los síntomas. Mientras tanto, existe una opción de tratamiento específica en la que la enzima que falta se sustituye por una enzima producida biotecnológicamente. La alfa-galactosidasa A producida en el laboratorio se administra al paciente mediante infusión y garantiza la descomposición del material de almacenamiento acumulado. Sin embargo, dado que la terapia de sustitución enzimática no puede reparar el daño orgánico que ya se ha producido, sino sólo retrasar su progresión, debe utilizarse lo antes posible. Otra opción es tomar una chaperona farmacológica por vía oral. Este agente puede utilizarse en pacientes con ciertas mutaciones en el gen GLA y actividad residual de la enzima α-galactosidasa A. Esto se debe a que se une a las formas inestables de AGAL y estabiliza la enzima para que pueda descomponer las sustancias grasas acumuladas en la célula.
Fuente: CardioMedLive 2020
CARDIOVASC 2020; 19(3): 28-29 (publicado el 17/9/20, antes de impresión).