Con el envejecimiento de la sociedad, las enfermedades de SMD también van en aumento. Sin embargo, los avances en el diagnóstico molecular y las nuevas terapias también cambiarán la gestión de los pacientes de más edad. Un enfoque multidisciplinar y el intercambio son importantes.
Los síndromes mielodisplásicos (SMD) se diagnostican como enfermedades de la tercera edad principalmente en pacientes >70 años. En Suiza, con una incidencia de 2-3/100.000 pacientes-año, cabe esperar algo más de 300 nuevos casos al año. Se calcula que en la actualidad viven en nuestro país unos 1600 pacientes con SMD [1]. Las opciones terapéuticas han permanecido esencialmente sin cambios en los últimos años, con el trasplante alogénico de células madre como tratamiento curativo posible sólo para unos pocos pacientes, y diversas opciones para mejorar las citopenias en el resto de situaciones paliativas [2–5]. En cambio, el rápido desarrollo de la “secuenciación de próxima generación” (NGS) también ha dado lugar a nuevos hallazgos relevantes en la rutina clínica de la hematología [6,7] que desempeñan un papel para los pacientes con SMD en lo que respecta al diagnóstico y la evaluación del pronóstico. Basándose en la clasificación actualizada de la OMS de 2016, a continuación se presentarán los avances y conceptos más importantes.
Revisión de la Clasificación de la OMS 2016
Incluso en la era de la biología molecular, la evaluación morfológica de los frotis de sangre periférica y los aspirados de médula ósea sigue siendo la base del diagnóstico. Básicamente, las formas de SMD con exceso de blastos se distinguen de las que carecen de proliferación blástica. El número de filas celulares afectadas por citopenias y displasias, la detección de sideroblastos en anillo (RS) y los cambios citogenéticos típicos son decisivos para la subdivisión posterior. Una novedad de la clasificación de la OMS de 2016 es la nomenclatura (Tab. 1) [8,9]. Los términos “anemia refractaria” o “citopenia refractaria” se han abandonado y ahora se utiliza el término “síndrome mielodisplásico” para todas las entidades, complementado con el hallazgo morfológico central. Esto aclara algunas incongruencias de la terminología anterior, como el término “anemia refractaria” para las formas de SMD con exceso de blastos que suelen ir acompañadas de pancitopenia.

La citogenética metafásica convencional sigue siendo el segundo pilar indispensable del diagnóstico de los SMD. En pacientes con citopenias sin proliferación blástica y sin displasias (significativas), el diagnóstico puede realizarse mediante la detección de ciertas anomalías citogenéticas definitorias de SMD (Tab. 2). En este caso, se asigna el diagnóstico “SMD inclasificable”. Esta categoría incluye también los casos con pancitopenia y displasia unilineal o con detección constante de blastos del 1% en la sangre periférica sin multiplicación de blastos en la médula ósea [8,9].
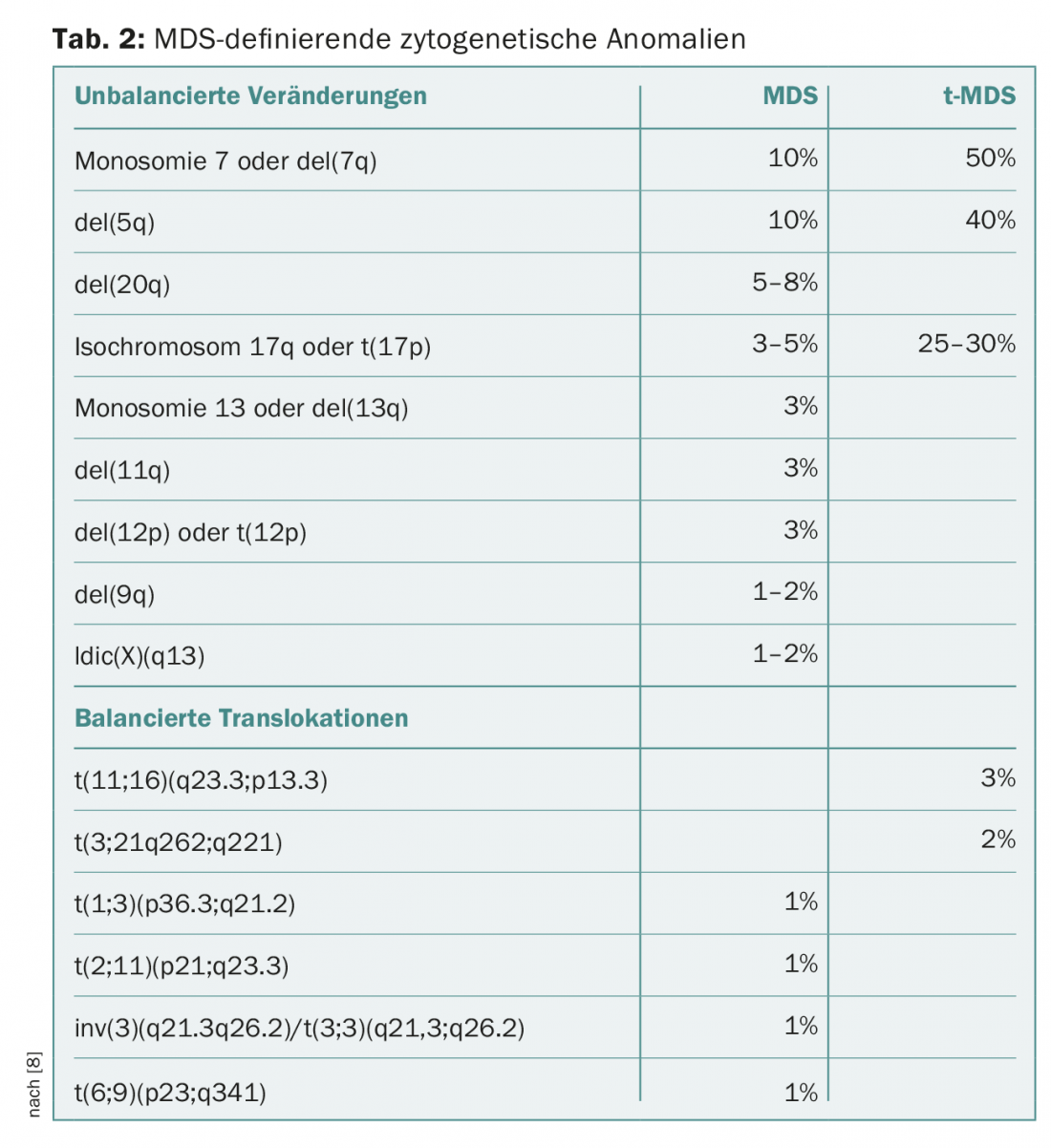
La categoría de SMD caracterizados por una deleción en el brazo corto del cromosoma 5 con del(5q) se ha ampliado en su definición. Ahora bien, los casos con una segunda anomalía citogenética también pueden asignarse a esta categoría, salvo en el caso de anomalías adicionales en el cromosoma 7, que se asocian a un pronóstico significativamente peor [8–10].
La citogenética convencional (se requiere el análisis de al menos 20 metafases para un examen concluyente) puede complementarse con otros métodos en situaciones seleccionadas, por ejemplo, en caso de una alta sospecha morfológica de SMD del(5q) pero cariotipo normal. Pueden utilizarse paneles FISH centrados en las anomalías cromosómicas típicas de los SMD o microarrays de hibridación genómica comparativa de todo el genoma (array-CGH) [11]. Sin embargo, estos dos métodos no ofrecen una sustitución a priori de la citogenética convencional. En particular, aún no se ha aclarado con certeza la importancia pronóstica de las anomalías que sólo pueden detectarse en el array CGH.
Importancia diagnóstica de la secuenciación de “próxima generación” en citopenias poco claras
En los últimos años, se han identificado numerosas mutaciones en genes asociados al desarrollo de SMD y a la progresión a LMA. El espectro de “genes impulsores” mutados es diverso y a menudo incluye componentes de la modificación epigenética del ADN y las histonas o la maquinaria de empalme modificadora del ARN (“espliceosoma”). También pueden verse afectados componentes de la regulación del ciclo celular, complejos de cohesina, factores de transcripción o componentes de la transducción de señales intracelulares (Tab. 3) [2,12–18].
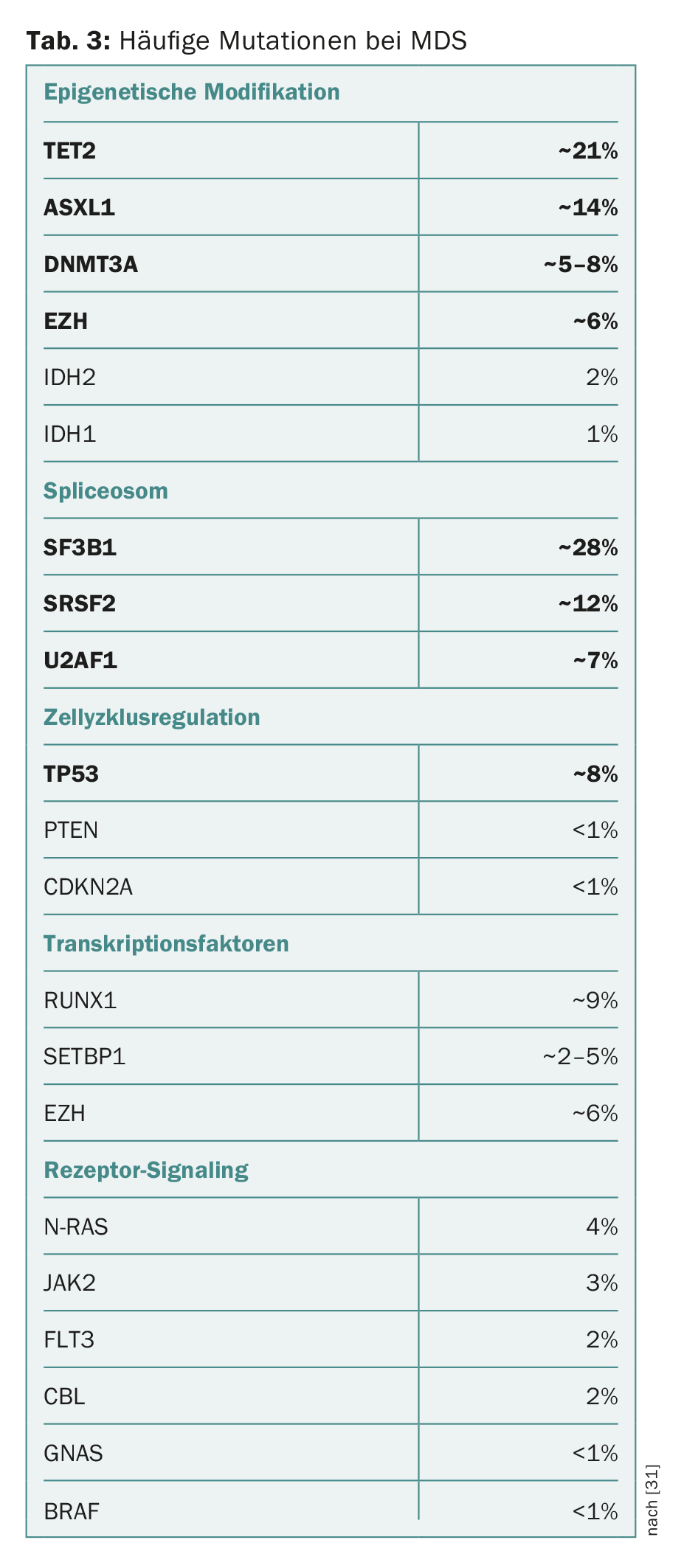
Por un lado, cabe destacar que la mayoría de las mutaciones de genes controladores encontradas en los SMD también se dan en otras neoplasias mieloides, aunque en frecuencias o combinaciones diferentes. Además, varios estudios han demostrado que las mutaciones típicas de las neoplasias mieloides también pueden darse en individuos hematológicamente sanos. La incidencia aumenta bruscamente con la edad, del 10% en personas de 60 años al 15-20% en >personas de 80 años (pero <1% en <personas de 40 años). Este fenómeno se ha denominado “hematopoyesis clonal de potencial indeterminado” (CHIP) [19,20]. Al igual que la gammapatía monoclonal de significado desconocido (GMSI) y la linfocitosis monoclonal de células B (LMBB), se trata de una afección precancerosa facultativa que puede progresar a enfermedad hematológica maligna a un ritmo aproximado del 1% anual. Además, los pacientes con CHIP también presentan una mayor morbilidad cardiovascular [21].
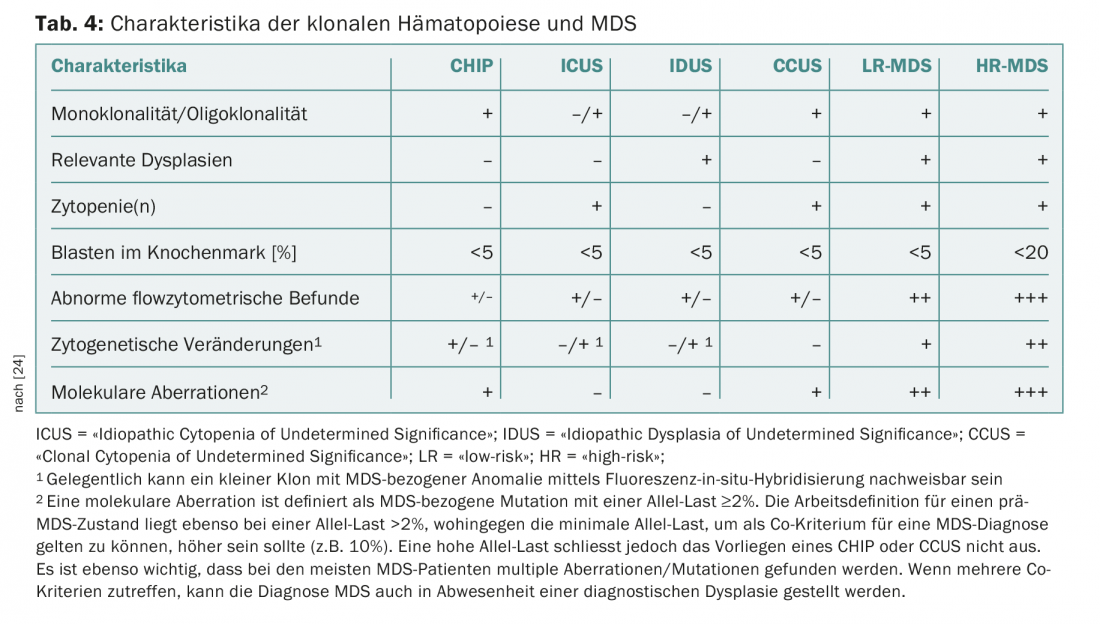
Un problema recurrente en la consulta y la clínica son los pacientes con citopenias persistentes para las que no se encuentran causas definidas. En ausencia de displasia o de anomalías citogenéticas definitorias de SMD, hasta ahora se han agrupado bajo el término “citopenia idiopática de significado indeterminado” (CIUS) [22]. La detección de mutaciones recurrentes puede ayudar a distinguir las citopenias reactivas de las clonales. La interpretación de la detección de una mutación depende del tamaño del clon (medido como “frecuencia alélica variante”, FAV) y del número de mutaciones detectadas. Si en una constelación de ICUS se encuentra una mutación recurrente con un VAF superior al 2%, se habla de “citopenia clonal de significación indeterminada” (CCUS). Entre los pacientes con CCUS, aquellos con al menos dos mutaciones con un VAF >10% tienen un alto riesgo de desarrollar una neoplasia hematológica en los próximos cinco años [23]. Los criterios presentados recientemente por un panel internacional de expertos para distinguir el CHIP, el ICUS y el CCUS del SMD manifiesto [24] se resumen en la tabla 4. Este consenso también define nuevos criterios menores relacionados con los SMD, que pueden utilizarse para un diagnóstico provisional de SMD en situaciones no concluyentes (Tab. 5).

La acumulación secuencial de alteraciones genéticas en las CEH se ha postulado durante mucho tiempo como un correlato patogenético de la evolución clonal, y el concepto de hematopoyesis clonal demuestra un solapamiento entre las alteraciones de la hematopoyesis con la edad y la patogénesis de las neoplasias mieloides. Sin embargo, sigue sin estar claro qué factores son responsables de la transición de CHIP y CCUS a neoplasia manifiesta. La evolución clonal parece estar causada no sólo por mecanismos intrínsecos a la célula (mutaciones en las células madre hematopoyéticas), sino también por mecanismos extrínsecos a la célula. A este respecto, el microentorno de la médula ósea y los componentes del sistema inmunitario innato y adquirido desempeñan un papel. El “estrés inmunológico” probablemente explica también la asociación con enfermedades o fenómenos inflamatorios e inmunológicos concomitantes, algunos de los cuales no pueden clasificarse con mayor precisión, que pueden darse en pacientes con SMD [25–28]. La pérdida del control inmunológico del tumor, así como los cambios facilitadores en el nicho de la médula ósea, son actualmente el centro de la investigación básica. En el futuro, esto podría dar lugar a nuevos enfoques terapéuticos que podrían utilizarse en una fase temprana del desarrollo de las neoplasias mieloides (Fig. 1).
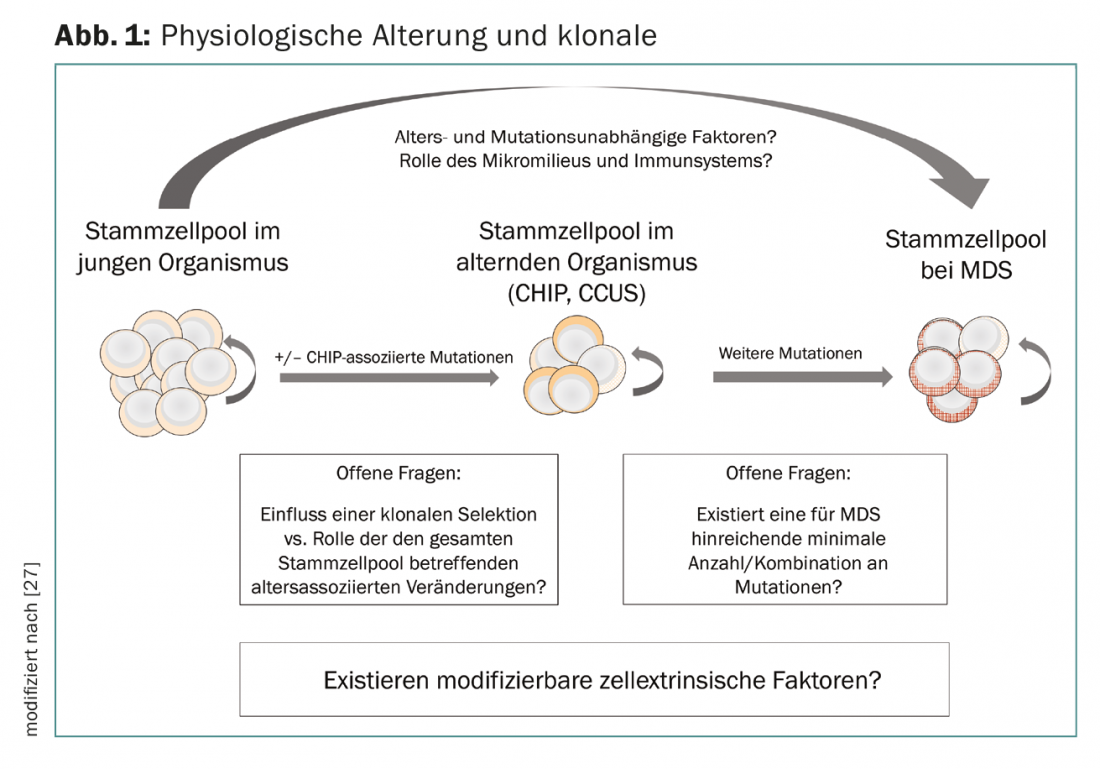
Debido al solapamiento del espectro de mutaciones entre el CHIP, el CCUS y el SMD, los análisis de mutaciones no se incluyeron deliberadamente en la clasificación actual de la OMS. Una excepción son las mutaciones en el gen conductor del componente del espliceosoma SF3B1, que están muy fuertemente asociadas a un fenotipo ringsideroblástico [29]. Según la OMS 2016, en el caso de una mutación SF3B1, la detección de un 5% de SR es suficiente para la clasificación en el grupo de SMD con SR, en lugar del 15% que se requiere en otras circunstancias. Los pacientes con mutación de SF3B1 tienen un pronóstico muy bueno con baja probabilidad de progresión a LMA. Dado que los pacientes con displasia multilinaje y SR también se benefician de la influencia pronóstica de una mutación SF3B1, se reintrodujo la entidad SMD con displasia multilinaje y SR en la clasificación de 2016.
Mayor importancia pronóstica y predictiva de las mutaciones de los genes impulsores
Desde un punto de vista pronóstico, ya se pueden nombrar algunos otros escenarios clínicos en los que los análisis de mutaciones pueden aportar información relevante. Alrededor del 15% de los pacientes con SMD del(5q) presentan una mutación TP53. Aunque éstos también responden hematológicamente a la lenalidomida, tienen menos probabilidades de alcanzar la remisión citogenética y un mayor riesgo de transición a la LMA. Por lo tanto, si se detecta una mutación TP53 en el SMD del(5q), pueden considerarse terapias alternativas [3,30].
Otro escenario para la búsqueda de mutaciones se refiere al grupo heterogéneo de pacientes en el grupo “intermedio” según la IPSS-R. Dependiendo de la presencia o ausencia de otras características de riesgo, estos pacientes pueden ser tratados según las recomendaciones aplicables a los pacientes de “bajo” o “alto riesgo” [2–5,30]. Hasta ahora, sólo se dispone para este fin de marcadores de riesgo convencionales (por ejemplo, elevación de LDH, fibrosis de médula ósea >grado 2 según la OMS) además del examen aislado de la citogenética (¿constelación de alto riesgo?). Varias mutaciones recurrentes se asocian a un riesgo significativamente mayor y justifican la elevación pronóstica a la categoría de “alto riesgo” y, en pacientes seleccionados, el trasplante alogénico de células madre [15]. Por ello, la búsqueda de mutaciones en TP53, ASXL1, RUNX1, EZH2 y ETV6 en pacientes que cumplen los requisitos para recibir terapia intensiva se recomienda explícitamente en las directrices actuales [4,15]. Un proyecto internacional patrocinado por la Fundación SMD se esfuerza actualmente por desarrollar un “IPSS-R molecular” complementado por el estado de mutación.
Los datos clínicamente útiles sobre el valor predictivo del perfil de mutaciones en los SMD son actualmente limitados. En el futuro se perfila la importancia de la estratificación con respecto a las terapias dirigidas. El luspatercept (ACE-356) es un inhibidor de la superfamilia TGF-β [10], que ha demostrado una elevada tasa de respuesta eritroide en pacientes con SMD refractarios a la EPO con RS y/o mutaciones en SF3B1. Además, actualmente se están investigando inhibidores del espliceosoma (H3B-8800) en la LMA y los SMD que, debido a la haploinsuficiencia, eliminan preferentemente los clones con mutaciones en los genes impulsores del espliceosoma (efecto cíclope). Además, las sustancias midostaurina (mutaciones FLT3), enasidenib (mutaciones IDH2) e ivosidenib (mutaciones IDH1), que ya han sido aprobadas para la LMA, también se encuentran actualmente en fase de desarrollo clínico para los SMD de alto riesgo con un perfil de mutación correspondiente, ya sea como sustancia única o en combinación con la terapia hipometilante (HMT) o la quimioterapia estándar.
Mensajes para llevarse a casa
- Debido al envejecimiento de nuestra sociedad, cabe esperar un aumento significativo de la enfermedad de SMD.
- Los avances en el diagnóstico molecular, así como las nuevas opciones terapéuticas, cambiarán también las estrategias de tratamiento para los pacientes de más edad.
- La gestión multidisciplinar es un requisito previo importante y plantea nuevos retos a los sistemas sanitarios. Esto afecta no sólo a la atención adecuada en la práctica clínica habitual, sino también a la realización de ensayos clínicos, que requieren un alto grado de cooperación y coordinación en el caso de las enfermedades raras en la era de la medicina personalizada.
- En respuesta a estos retos, el Grupo de Estudio Suizo de SMD lanzó en 2015 el Registro/Biobanco Suizo de SMD para facilitar el intercambio clínico y científico dentro de una red internacional.
Literatura:
- Bonadies N, et al: Tendencias de clasificación, incidencia, mortalidad y supervivencia de los pacientes con SMD en Suiza entre 2001 y 2012. epidemiología del cáncer 2017; 46: 85-92.
- Malcovati L, et al: Diagnóstico y tratamiento de los síndromes mielodisplásicos primarios en adultos: Recomendaciones de la European LeukemiaNet. Sangre 2013; 122(17): 2943-2964.
- Fenaux P, et al: Síndromes mielodisplásicos: Guía de práctica clínica de la ESMO para el diagnóstico, tratamiento y seguimiento. Anales de oncología: revista oficial de la Sociedad Europea de Oncología Médica 2014; 25(Suppl 3): iii57-69.
- Hofmann WK, Platzbecker U, Götze K: Onkopedia-Leitlinie Myelodysplastische Syndrome. Estado marzo 2016.
- Greenberg PL, et al: Síndromes mielodisplásicos, versión 2.2017, Guías de práctica clínica en oncología de la NCCN. Revista de la Red Nacional Integral del Cáncer: JNCCN 2017; 15(1): 60-87.
- Johnsen JM, Nickerson DA, Reiner AP: Secuenciación masiva en paralelo: la nueva frontera de la genómica hematológica. Sangre 2013; 122(19): 3268-3275.
- Kuo FC, et al: The relative utilities of genome-wide, gene panel, and individual gene sequencing in clinical practice. Sangre 2017; 130(4): 433-439.
- Swerdlow SH, et al. (Eds.): Clasificación de la OMS de los tumores de los tejidos hematopoyéticos y linfoides. 4ª edición revisada. Lyon: Centro Internacional de Investigaciones sobre el Cáncer 2017.
- Arber DA, et al: La revisión de 2016 de la clasificación de la Organización Mundial de la Salud de las neoplasias mieloides y la leucemia aguda. Sangre 2016; 127(20): 2391-2405.
- Mies A, Platzbecker U: Aumento de la eficacia de la hematopoyesis en los síndromes mielodisplásicos: agentes estimulantes de la eritropoyesis e inhibidores de la superfamilia del factor de crecimiento transformante-β. Seminarios de hematología 2017; 54(3): 141-146.
- Ouahchi I, et al.: La hibridación genómica comparativa basada en micromatrices revela aberraciones recurrentes adicionales en pacientes adultos evaluados por síndrome mielodisplásico con cariotipo normal. Revista británica de hematología 2018. DOI: 10.1111/bjh.15068 [Epub ahead of print].
- Kon A, et al.: Mutaciones recurrentes en múltiples componentes del complejo cohesina en neoplasias mieloides. Nature genetics 2013; 45(10): 1232-1237.
- Yoshida K, et al.: Frecuentes mutaciones de la vía de la maquinaria de splicing en la mielodisplasia. Naturaleza 2011; 478(7367): 64-69.
- Bejar R, Levine R, Ebert BL: Desentrañando la fisiopatología molecular de los síndromes mielodisplásicos. Revista de oncología clínica: revista oficial de la Sociedad Americana de Oncología Clínica 2011; 29(5): 504-515.
- Bejar R, et al.: Efecto clínico de las mutaciones puntuales en los síndromes mielodisplásicos. The New England journal of medicine 2011; 364(26): 2496-2506.
- Abdel-Wahab O, Figueroa ME: Interpretación de la nueva genética molecular en los síndromes mielodisplásicos. Hematología Programa educativo de la Sociedad Americana de Hematología 2012; 2012: 56-64.
- Leeke B, et al.: Mutaciones de la cohesina en neoplasias mieloides: Mecanismos subyacentes. Hematología y oncología experimentales 2014; 3: 13.
- Tothova Z, Steensma DP, Ebert BL: Nuevas estrategias en los síndromes mielodisplásicos: Aplicación del diagnóstico molecular a la práctica clínica. Clinical cancer research: an official journal of the American Association for Cancer Research 2013; 19(7): 1637-1643.
- Jan M, Ebert BL, Jaiswal S: Hematopoyesis clonal. Seminarios de hematología 2017; 54(1): 43-50.
- Heuser M, Thol F, Ganser A: Hematopoyesis clonal de potencial indeterminado. Deutsches Arzteblatt internacional 2016; 113(18): 317-322.
- Fuster JJ, Walsh K: Mutaciones somáticas y hematopoyesis clonal: nuevos impulsores potenciales inesperados de las enfermedades cardiovasculares relacionadas con la edad. Circulation research 2018; 122(3): 523-532.
- Valent P, et al.: Citopenia idiopática de significado indeterminado (ICUS) y displasia idiopática de significado incierto (IDUS), y su distinción de los SMD de bajo riesgo. Investigación sobre la leucemia 2012; 36(1): 1-5.
- Malcovati L, et al.: Importancia clínica de la mutación somática en la citopenia sanguínea inexplicada. Sangre 2017; 129(25): 3371-3378.
- Valent P, et al: Propuesta de criterios diagnósticos mínimos para los síndromes mielodisplásicos (SMD) y posibles afecciones previas a los SMD. Oncotarget 2017; 8(43): 73483-73500.
- Gañán-Gómez I, et al.: Desregulación de la señalización inmunitaria innata e inflamatoria en los síndromes mielodisplásicos. Leucemia 2015; 29(7): 1458-1469.
- Glenthøj A, et al: Mecanismos inmunitarios en el síndrome mielodisplásico. Revista internacional de ciencias moleculares 2016; 17(6): 944.
- Chung SS, Park CY: Envejecimiento, hematopoyesis y los síndromes mielodisplásicos. Avances en sangre 2017; 1(26): 2572-2578.
- Cooper JN, Young NS: Clonalidad en contexto: Clones hematopoyéticos en su entorno de médula. Sangre 2017; 130(22): 2363-2372.
- Malcovati L, et al: La mutación SF3B1 identifica un subconjunto distinto de síndrome mielodisplásico con sideroblastos en anillo. Sangre 2015; 126(2): 233-241.
- Grupo Nórdico de Estudio de los SMD: Directrices para el tratamiento de los pacientes con síndromes mielodisplásicos y leucemia mielomonocítica crónica. 8ª actualización. www.nmds.org/index.php/guidelines (citado el 14.03.2018).
- Montalbán-Bravo G, García-Manero G: Síndromes mielodisplásicos: actualización de 2018 sobre diagnóstico, estratificación del riesgo y manejo. Am J Hematol 2018 Jan; 93(1): 129-147.
Para saber más:
- List A, Ebert BL, Fenaux P: Una década de progresos en el síndrome mielodisplásico con deleción del cromosoma 5q. Leucemia 2018. DOI: 10.1038/s41375-018-0029-9 [Epub ahead of print].
- Platzbecker U, et al: Luspatercept para el tratamiento de la anemia en pacientes con síndromes mielodisplásicos de bajo riesgo (PACE-MDS): Un estudio multicéntrico, abierto de fase 2 de búsqueda de dosis con estudio de extensión a largo plazo. The Lancet Oncology 2017; 18(10): 1338-1347.
InFo ONCOLOGÍA & HEMATOLOGÍA 2018; 6(2): 22-26.









